登录/注册后可看大图
北京时间12月11日,百济神州在佐治亚州亚特兰大举行的第59届美国血液学协会(ASH)年会上发布了其在研布鲁顿氏酪氨酸激酶(BTK)抑制剂zanubrutinib (BGB-3111)与在研抗PD-1抗体tislelizumab (BGB-A317)联合用于B细胞恶性肿瘤患者的1b期临床试验(进行中)的初始数据。剂量递增的初始数据表明,zanubrutinib与tislelizumab联合用药在B细胞恶性肿瘤患者中具有可控的毒性特征和抗肿瘤活性。
澳大利亚Peter MacCallum癌症研究中心低度淋巴瘤和慢性淋巴细胞白血病疾病小组负责人、St. Vincent医院血液学主任兼报告第一作者C**tantine **医学博士表示: “本项1b期试验的初始数据表,zanubrutinib与tislelizumab联合用药是可以耐受的;不良事件与各治疗类别大致一致。尽管随访时间较短,我们已经在既往接受过多次治疗的不同恶性肿瘤患者人群中观察到了客观缓解。”
百济神州血液学首席医疗官黄蔚娟医学博士说道:“临床前数据表明该联合用药具有协同作用,我们希望本项临床试验将展示该联合用药对B细胞恶性肿瘤患者,尤其是侵袭性淋巴瘤患者的治疗潜力。”
正在进行的1b期临床试验的结果总结
Zanubrutinib与tislelizumab联合用药的开放性、多中心1b期临床试验包括剂量递增部分和随后的剂量扩展部分。在ASH上发表的数据包括在剂量递增期前两个剂量水平下招募的患者:剂量1组为zanubrutinib 320mg每日一次(QD)与tislelizumab 2mg/kg每3周一次(Q3W),剂量2组为zanubrutinib 320mg QD与tislelizumab 5mg/kg Q3W。第3个剂量组中的患者将接受zanubrutinib 160mg每日两次与tislelizumab 200mg Q3W的联合治疗。
截至最新数据截点2017年9月15日,25名患者包括剂量1组中的15名患者和剂量2组中的10名患者已入组本试验。其中13名为惰性淋巴瘤患者,包括慢性淋巴细胞白血病 (CLL)、滤泡性淋巴瘤(FL)、边缘区淋巴瘤(MZL)和华氏巨球蛋白血症(WM)。另外12名为侵袭性淋巴瘤患者,包括弥漫性大B细胞淋巴瘤(DLBCL)、 套细胞淋巴瘤(MCL)和转化型淋巴瘤。中位随访时间为5.1个月(0.4-14.1个月)。在剂量2组的WM患者中发生2例自身免疫性溶血,且其中1例可视为剂量限制性毒性(DLT)。这些事件与直接抗球蛋白试验阳性无关,并经免疫抑制剂治疗得到缓解,但因此决定在本项试验中排除WM患者的进一步招募。排除WM患者后,未观察到其他DLT。
在惰性淋巴瘤患者中,最常见的任何归因不良事件(AE)(发生在20%及以上的患者中)是瘀斑/紫癜/挫伤(31%)和血小板减少症(23%)。发生在至少2名患者中的任何归因的3-4级AE包括血小板减少症、贫血和溶血(各15%)。除2例自身免疫溶血事件外,另外还发生1例免疫相关事件即4级自身免疫性脑炎。该患者随后接受了强效免疫抑制治疗并逐渐恢复。
在侵袭性淋巴瘤患者中,最常见的任何归因的AE(发生在20%及以上患者中)是腹泻、疲劳、发热、上呼吸道感染(各33%)、咳嗽(25%)和恶心(25%)。发生在至少2名患者中的3-4级任何归因AE包括发热(17%)。有一名患者报告了多次2级和3级非感染性肺炎。
到数据截点为止,25名患者满足疗效评估条件。中位随访时间为5.1个月(0.4-14.1个 月)。在10名患者(40%)中观察到客观缓解。按肿瘤类型分类,在5名CLL患者中观察到2例部分缓解(PR),在5名FL患者中观察到1例完全缓解(CR)和1例PR, 在2名WM患者中观察到1例非常好的部分缓解和1例轻度缓解,在5名DLBCL患者中观察到1例CR,以及在5名转化型淋巴瘤患者中观察到3例PR。
Omeros公司是一家商业化的生物制药公司,一直致力于探索、开发和销售针对大型市场的小分子和蛋白质疗法药物,还有罕见病药物如炎症、补体介导的中枢神经系统疾病和紊乱。近日,该公司宣布FDA在审查了药物OMIDRIA儿科临床试验的有效性和安全性数据之后,批准了该药物的补充性新药申请(sNDA),同意将OMIDRIA(去氧肾上腺素和酮咯酸眼内溶液)的适应症扩大至应用于儿科患者。
2014年,FDA已批准其眼科药物Omidria用于白内障和晶状体置换术,这也是该公司开发并获得药品监管部门上市许可的首款药物。
该药物成功地在78名儿科患者中进行了临床试验,随机分配成两组,进行OMIDRIA或苯肾上腺素术中。除了已获授权的儿科和**患者适应症之外,FDA还授予OMIDRIA额外6个月在美国地区市场的独家经营权。根据“联邦食品药品和化妆品法案”第505A节,这项额外6个月的市场独占权延期与FDA“橙皮书”中列出的药品专利期限有关。
对这项喜人的结果,Omeros董事长兼首席执行官Gregory A. Demopulos表示:“我们很高兴地得知FDA批准在儿科患者中使用OMIDRIA并获得额外的市场独占权。现在,不管是**还是儿童,所有接受白内障或其他晶状体置换手术的患者都可以获得OMIDRIA的治疗机会。药物审批程序在儿科患者中比在**中要来得更为复杂,未来我们的药物将帮助更多的儿童患者,这是非常令人高兴的一件事。”
Omeros此前宣布与Par Pharmaceutical,Inc.及其子公司Par Sterile Products,LLC(Par)的专利侵权诉讼获得了有利的解决,Par在其中充分知晓并确认了橙皮书中OMIDRIA的专利有效性。除非事先根据和解协议中的条款获得授权,否则Par将被禁止在2032年4月1日之前发布任何OMIDRIA的仿制药品。
同样,这些专利诉讼也存在于Omeros与Sandoz和Lupin有限公司之间,这个诉讼计划在2019年年中在美国特拉华州地区法院进行审判。如果该法院支持并认定在2033年10月到期的OMIDRIA橘皮书上市专利有效,由于FDA新授予OMIDRIA额外6个月的市场专有权,Sandoz和Lupin两家公司将被禁止发布OMIDRIA的仿制版本,直到2034年4月。
7、FDA今**准罕见血管炎首个新药
美国FDA今天宣布批准葛兰素史克(GSK)的Nucala(mepolizumab)扩大应用范围,用以治疗患嗜酸性**肿性血管炎(EGPA)的成年患者。EGPA是一种罕见的自身免疫性疾病,可导致血管炎。Nucala是第一个FDA批准的专门治疗EGPA的药物。此外,FDA授予Nucala孤儿药资格。
根据美国国立卫生研究院(NIH)的统计,EGPA(以前称为Churg-Strauss综合征)这种罕见病的病征包括哮喘、高水平嗜酸性粒细胞(一种有助于抵抗感染的白血细胞)和中、小血管炎症。发炎的血管可以影响各种器官系统,包括肺、胃肠道、皮肤、心脏和神经系统。据估计,每100万人每年约有0.11至2.66个新病例,累计每100万成年人总体患病率为10.7至14。
Nucala是由重组DNA技术生产的抗白细胞介素-5的单克隆抗体。Nucala之前在2015年被批准用于治疗12岁以上的有特殊情况的哮喘患者,这些患者尽管已经接受了哮喘药物治疗,但依然表现严重哮喘并伴嗜酸性表型。
Nucala的安全性和有效性基于52周MIRRA临床试验的数据,该试验检验Nucala和安慰剂相比的疗效。患者每四周一次接受300毫克Nucala或安慰剂皮下注射,同时进行常规每日口服皮质类固醇(OCS)治疗。从第四周开始,逐渐减少OCS剂量。在OCS剂量减小至小于或等于4毫克泼尼松时,评估测量Nucala的治疗对疾病缓解的影响。结果显示,与安慰剂相比,接受Nucala的患者获得缓解的积累时间显著延长。在治疗的第36周和48周,接受Nucala的患者缓解比例显著高于安慰剂组。另外,与安慰剂组相比,接受Nucala的患者在治疗的第24周内达到缓解的比例显著提高,并且在52周治疗研究期间持续获得缓解。
美国FDA药物评估和研究中心的肺、过敏和风湿病产品部主任Badrul Chowdhury博士说:“在今天之前,患有这种具有挑战性的罕见疾病的患者没有FDA批准的治疗方案。扩大的Nucala的适应症满足了EGPA患者的一个关键、未获满足的需求。值得注意的是,在临床试验中服用Nucala的患者的症状有明显的改善。”
宾夕法尼亚大学佩雷尔曼医学院风湿病学系主任,参与此项MIRRA临床研究的Peter A. Merkel博士说:“患有EGPA的患者往往面临一个令人沮丧的旅程,从接受正确诊断的延迟到少有安全并有效的治疗方案。风湿病学家、免疫学家和肺科医生在正确诊断和治疗EGPA患者中起着重要的作用。今天批准的mepolizumab为专家提供了针对这种复杂疾病的合适患者提供针对性治疗的能力。”
MIRRA研究的首席研究员Michael E. Wechsler博士说:“EGPA患者经常忍受反复的复发,使他们的组织和器官承担更高的永久性损伤风险。临床资料表明,mepolizumab增加了缓解时间,减少了复发,使患者与安慰剂组相比可以使用更低剂量的皮质类固醇。这些是关键的治疗目标,这一批准对医生和病人来说都是一个重要的里程碑。”
我们祝贺Nucala获准应用于EGPA这一难治疾病的治疗,也为更多患者能够因此得到缓解而感到欣慰。
参考资料:
[1] FDA approves first drug for Eosinophilic Granulomatosis with Polyangiitis, a rare disease formerly known as the Churg-Strauss Syndrome
[2] GSK achieves approval for Nucala (mepolizumab) for the treatment of eosinophilic granulomatosis with polyangiitis (EGPA) for adults in the US
8、拜耳和强生提交利伐沙班CAD/PAD新适应症
德国拜耳与合作伙伴美国强生近日联合宣布,双方已向美国食品和药物管理局(FDA)提交了口服抗凝血剂Xarelto(品牌名:拜瑞妥,通用名:rivaroxaban,利伐沙班)的一份补充新药申请(sNDA)。该sNDA寻求FDA批准利伐沙班(2.5mg,每日2次)联合低剂量阿司匹林(100mg,每日一次),用于2个新的适应症:(1)用于冠状动脉和/或外周动脉疾病(CAD/PAD)患者,降低主要不良心血管事件(MACE)风险,包括CV死亡、心脏病发作及中风;(2)用于PAD患者,降低急性肢体缺血风险。
此次sNDA的提交,是基于III期临床研究COMPASS的数据。该研究是迄今为止调查利伐沙班疗效和安全性的最大规模临床研究,由拜耳和强生与加拿大人口健康研究所(PHRI)合作开展,共入组了全球30多个国家600多个网点27395例患者。研究中,患者以1:1:1的比例随机分配接受利伐沙班(2.5mg,每日2次)+阿司匹林(100mg,每日一次)联合治疗、利伐沙班(5mg,每日2次)单药治疗、阿司匹林(100mg,每日一次)单药治疗。入组研究前接受质子泵抑制剂(PI)治疗的患者将继续使用现有药物,未接受PI的患者将随机接受泮托拉唑(pantoprazole)或安慰剂。研究的目的是评估利伐沙班用于CAD、PAD及CAD/PAD患者预防MACE(包括CV死亡,心肌梗死[MI]和中风)的疗效和安全性。
今年2月,由于卓越的疗效,COMPASS研究比原计划提前大约一年时间达到主要终点,并根据独立数据监测委员会(DMC)建议已提前终止。COMPASS研究主要数据:
主要疗效结果:
(1)预防中风、CV死亡和MI复合终点方面,利伐沙班(2.5mg,每日2次)+阿司匹林(100mg,每日一次)组合疗法显著优于利伐沙班(5mg,每日2次)单药疗法和阿司匹林(100mg,每日一次)单药疗法(HR=0.76,95%CI:0.44-0.76,p<0.001)。
(2)利伐沙班(2.5mg,每日2次)+阿司匹林(100mg,每日一次)组合疗法使中风、CV死亡、心脏病发作风险分别降低42%(HR=0.58,95%CI=0.44-0.76,p<0.001)、22%(HR=0.78,95%CI=0.64-0.96,p=0.02)、14%(HR=0.86,95%CI=0.70-1.05,p=0.14),利伐沙班(5mg,每日2次)单药疗法也降低了中风、CV死亡、MI复合终点,但结果不具有统计学显著差异。
(3)与阿司匹林(100mg,每日一次)单药疗法相比,利伐沙班(2.5mg,每日2次)+阿司匹林(100mg,每日一次)组合疗法也显著改善了净临床受益(HR=0.80,95%CI=0.70-0.91,p<0.001;净临床受益定义为中风、CV死亡、MI、重要器官致死性出血或症状性出血),利伐沙班(5mg,每日2次)单药疗法与阿司匹林(100mg,每日一次)单药疗法相比未能改善净临床受益。
安全性结果:主要安全性预后是修订版的国际血栓与止血学会(ISTH)大出血标准,包括致命性出血、重要器官的症状性出血、血液进入手术部位需要再次手术、出血导致住院治疗。数据显示,各个组出血发生率均很低;与阿司匹林(100mg,每日一次)单药疗法相比,尽管利伐沙班(2.5mg,每日2次)+阿司匹林(100mg,每日一次)组合疗法使大出血风险显著增加(HR=1.70,95%CI=1.40-2.05,p<0.001),但大部分大出血进入胃肠道,致死性出血、颅内出血、重要器官症状性出血无明显增加。重要的是,在PAD患者群体中,主要不良肢体事件以及所有由血管导致的截肢显著降低。
利伐沙班:全球应用最广泛的NOAC
利伐沙班是首个也是唯一一个被评估用于稳定/慢性CAD或PAD患者群体用于心血管疾病二级预防的非维生素K拮抗剂类口服抗凝剂(NOAC)。COMPASS研究是拜耳/强生利伐沙班广泛评估项目的一部分,除了该研究之外,拜耳也在评估利伐沙班用于心血管领域的其他研究,包括VOYAGER PAD和COMMANDER-HF。
CAD是导致心血管疾病的最常见原因,在全球范围内,每年有高达730万人死于CAD。在高收入国家中,多达1/3-1/2的中年男性和女性,在其一生中存在发生CAD的风险,而且全球CAD患者的数目也在不断攀升。截止2020年,CAD的治疗负担将达到820万伤残调整寿命年(DALYs)。而PAD,尽管确诊率低,但仍影响着欧洲和北美超过2700万人,是心血管疾病的一个重要风险标志。在全球范围内,筛查研究表明,55岁以上老年群体中,大约20%患有PAD。与CAD一样,PAD的发病率也与年龄密切相关,随着人口老龄化,患者的数目正在不断上升。
而利伐沙班将为CAD和PAD患者群体提供一个重要的治疗选择,用于降低其主要心脏不良事件风险。
利伐沙班目前已获批7个适应症,与其他NOAC相比,能够为广泛的患者群体提供帮助,预防多种静脉血栓栓塞(VTE)和动脉血栓栓塞(VAT)疾病疾病。利伐沙班由拜耳和强生合作开发,强生负责美国的销售,拜耳负责美国以外地区的商业化销售,已获全球130多个国家批准。
利伐沙班的适应症包括:(1)用于伴有一个或多个风险因素的非瓣膜性心房颤动(AF)**患者,预防卒中和全身性栓塞;(2)用于**患者肺栓塞(PE)的治疗;(3)用于**患者深静脉血栓(DVT)的治疗;(4)用于**患者预防PE和DVT复发;(5)用于择期髋关节置换术的**患者,预防VTE;(6)用于择期膝关节置换术的**患者,预防VTE;(7)在急性冠脉综合征(ACS)后心脏生物标志物升高且既往未发生卒中或短暂性脑缺血性发作(TIA)的**患者中,联合乙酰水杨酸(ASA)或ASA+一种噻吩并吡啶(氯吡格雷或噻氯匹定),预防动脉粥样硬化血栓**件(心血管死亡、心肌梗死、卒中)。(新浪医药编译/newborn)
文章参考来源:
[url=http://www.investor.bayer.de/index.php?id=145&L=1&tx_news_pi1[news]=2179]1、Bayer's rivaroxaban submitted to U.S. FDA for approval in patients with coronary and/or peripheral artery disease[/url]
[url=http://www.investor.bayer.de/index.php?id=145&L=1&tx_news_pi1[news]=1962]2、Phase III COMPASS study with Bayer's Rivaroxaban in Patients with Coronary or Peripheral Artery Disease Shows Overwhelming Efficacy and Meets Primary Endpoint Early[/url]
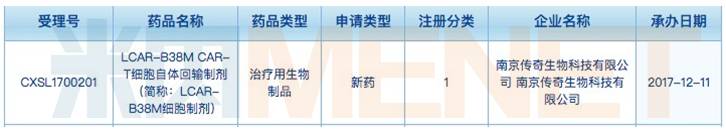
据CFDA药品审评中心12月11日披露,南京传奇生物科技有限公司(简称“南京传奇生物”)申报的LCAR-B38MCAR-T细胞自体回输制剂新药临床申请已被正式受理,这是我国首个获得受理的CAR-T疗法。
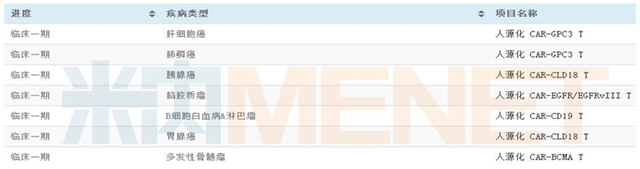
LCAR-B38M是南京传奇生物研发的一款靶向b细胞成熟抗原(BCMA)的CAR-T疗法。BCMA存在于成熟B细胞表面,属于TNF受体家族,是一种极为重要的B细胞生物标志物。南京传奇生物也凭借在CAR-T领域的突破性进展,在2017年ASCO年会(美国临床肿瘤协会年会)引起了诸多关注。
除去南京传奇生物,国内还有众多布局CAR-T疗法这一领域的企业。
CAR-T细胞治疗市场空间巨大
免疫系统是人体自身的医生,而肿瘤免疫治疗被认为是目前唯一有可能彻底治愈癌症的方法。CAR-T免疫疗法是肿瘤免疫治疗领域的“网红”,是当下最受关注的抗肿瘤技术之一。目前CAR-T细胞治疗多数被用于晚期肿瘤患者,但将来可能会像化疗一样,成为癌症治疗的一线方法。据Coherent Market Insights分析,CAR-T细胞治疗预计在2028年全球市场规模将达到85亿美元,未来的市场空间预计在350亿美元-1000亿美元之间。
竞争激烈,众多公司纷纷加入研究队伍
在CART-T疗法的开发上,美国遥遥领先,中国紧随其后,欧洲和日本大幅落后。美国是CAR-T技术的起源地,也是全球医药科技的龙头,各大医药巨头也纷纷涉足这一领域。其中诺华和Kite(已被吉利德收购)在CAR-T疗法的开发上处于领先地位,旗下的CAR-T技术各具特色,引领着CAR-T的潮流。另外还有多家小型生物医药公司也分别在各自的领域取得了一定突破,比如Cellectis(使用异体T细胞)、CBMG等。
中国此次也把握住了这一时代潮流,随着CAR-T细胞治疗在中国的研发快速发展,中国已然成为在CAR-T临床研究项目上与美国同列为全球CAR-T研究的第一梯队。据美国Clinical Trials.gov 网站对“Chimeric receptor”的搜索结果,截至目前中国登记开展CAR-T临床研究项目116项,已经在数量上超过欧洲,仅次于美国。其中西比曼、博生吉、合一康及宇研生物走在前列,2015年以后,复星医药、科济生物、安科生物、中源协和等公司也大手笔布局CAR-T细胞治疗。
国内研发领先的企业
复星医药:剑指国内CAR-T市场霸主地位
2017年1月11日,复星医药宣布,通过旗下全资子公司成立中外合营企业,拟在中国引进Kite Pharma公司(已被吉利德收购)的CAR-T治疗产品KTE-C19(商品名为Yescarta),为淋巴癌患者带来全球领先的治疗手段。KTE-C19于2017年10月18日获得FDA批准,是全球第二款获批的CAR-T疗法,对血液肿瘤有着显著的疗效。2017年12月5日,复星凯特生物科技有限公司(简称“复星凯特”)在上海张江举办了细胞治疗基地启动仪式暨CAR-T科学研讨会。作为复星医药和美国Kite Pharma的合营公司,复星凯特正在全面推进KTE-C19的技术转移、制备验证等工作,致力于早日推动该细胞治疗产品在国内的产业化。按照目前的进度,复星凯特的引进的KTE-C19极有可能是首款能在国内上市的CAR-T产品。
KTE-C19在米内网全球药物研发库的搜索结果
(点击图片可放大)
西比曼生物科技:来自中国,放眼世界
西比曼生物科技于2014年在美国纳斯达克挂牌上市,成为第一个在美国纳斯达克上市的中国细胞生物医药科技公司。西比曼的CAR-T临床研究主要与解放军301医院韩为东教授合作,2015年2月以1200万收购了解放军总医院(301医院)韩为东教授组的4项CAR-T 产品,并保留后续产品的优先收购权。西比曼在CAR-T疗法的临床试验主要集中在临床二期,从目前公司公布的临床试验结果来看,初步显示这些产品的相对安全、可行和有效。值得一提的是其针对实体瘤EGFR靶点的CAR-T疗法“CBM-EGFR.1”的一期临床数据,在多种类型实体瘤上取得了应答,研发进度处于国际领先水平。
西比曼的CAR-T产品研发管线
(来源:米内网全球药物研发库)
博生吉:老牌细胞治疗技术公司的厚积薄发
博生吉作为先行的国内免疫细胞疗法开发公司,主要业务为开发细胞治疗技术(CAR-T、CAR-NK、aAPCCTL和aAPCNK技术)和抗体靶向药物。2015年6月,安科生物出资2000万与博生吉强强联手,成立合资公司,专攻CAR-T细胞治疗技术研究及产业化运作。博生吉目前已经有3个CAR-T项目处于临床二期,其中包含了较难治愈的多种实体瘤,研发进度在国内处于领先位置。
博生吉的CAR-T产品研发管线
(来源:米内网全球药物研发库)
科济生物:专注于CAR-T疗法的创新企业
科济生物于2014年10月30日在上海成立,创新技术、多管齐下的研发管线以及经验丰富的创始团队,使得科济生物医药深受资本市场的青睐。早在2015年3月,科济就与上海仁济医院开展了CAR-T临床合作。2015年9月,科济生物与上海市肿瘤研究所就CAR-T等肿瘤免疫治疗签署了为期5年的战略合作协议。科济生物已开发出针对多种恶性实体肿瘤的CAR-T疗法,目前已将全世界第一个针对肝细胞肝癌(HCC)以及针对多形性脑胶质瘤(GBM)推上临床研究阶段。
科济生物的CAR-T产品研发管线
(来源:科济生物公司官网)
国内开发CAR-T疗法的优秀企业还有:药明巨诺、吉凯基因、恒润达生等公司,由于篇幅所限,在此不再一一叙述。
结语作为新兴的精准医学治疗手段,以CAR-T细胞治疗为代表的免疫疗法得到了快速发展,巨大的市场潜力吸引着诸多企业大手笔布局。前景虽然无限光明,但依旧存在如价格昂贵、无法规模批量生产、毒副作用(细胞因子释放综合征、神经系统毒性等)、对实体瘤效果不佳等诸多问题。不过相信随着医药研发者不断的努力,这些问题最终都能得到解决,让CAR-T疗法真正意义上地惠及众生。
3、和黄抗癌新药fruquintinib在美启动临床试验
今日,Hutchison China MediTech(Chi-Med,和黄中国医药科技)在美国启动了fruquintinib的1期桥接临床试验(bridging clinical trial)。
Fruquintinib(HMPL-013)是一款高度选择性血管内皮生长因子受体(VEGFR)1、2和3的口服抑制剂。与其它靶向治疗相比,通过口服剂量能24小时抑制VEGFR,且具有更低的脱靶毒性。其良好的耐受性,加上已被证明的药物相互作用安全性,使得它可以与其它癌症疗法进行合理组合,例如在正在进行的fruquininib的临床试验中,与化疗和其它靶向疗法组合。
在癌症的晚期阶段,肿瘤会分泌大量的蛋白质配体VEGF,以刺激肿瘤周围形成过度的血管生成,从而为肿瘤提供更多的血流、氧气和营养。VEGF和VEGFR在肿瘤相关的血管生成中起着关键作用,而fruquininib可以抑制VEGF/VEGFR通路。这是阻断肿瘤生长和侵袭所必需的新血管发展的重要治疗策略。
目前,正在中国开展的几项中后期临床试验在对fruquininib的疗效进行验证。一项关键3期临床试验FRESCO显示了fruquininib在416名晚期结直肠癌患者中的疗效,基于这一结果,CFDA于今年6月接受了fruquininib的新药申请,并授予其优先审评资格。一项包含了520名非小细胞肺癌(NSCLC)患者的关键3期临床试验FALUCA正在验证fruquininib治疗NSCLC的疗效。将fruquininib与Iressa(gefitinib,吉非替尼)联合用于一线治疗晚期或转移性NSCLC的研究正在2期临床阶段。此外,一项关键3期临床试验FRUTIGA正在超过500名晚期胃癌或胃食管连接处(GEJ)腺癌患者中验证fruquininib与Taxol(紫杉醇)联合治疗的效果。
相关阅读:8款主打新药,31项临床试验,和黄研发管线大盘点
此次在美国启动的研究是一项多中心、开放标签的1期临床试验,评估fruquintinib在美国晚期实体瘤患者中的安全性、耐受性和药代动力学。
我们期待这款新药能继续在临床试验中取得佳绩,为患者带来治疗新选择。
原标题 :祝贺!和黄抗癌新药在美国启动临床试验
参考资料:
[1] Chi-Med Initiates Fruquintinib U.S. Clinical Trials
[2] ASCO | 祝贺!和黄癌症新药3期成功,有望很快上市中国
4、我国首个用于肝癌二线治疗的新药获批
昨日(12月12日),拜耳对外宣布,其靶向抗肿瘤药物瑞戈非尼(商品名:拜万戈)被CFDA正式批准,用于既往接受过索拉非尼治疗的肝细胞癌(HCC)患者,这也是在华首个获批用于HCC二线治疗的新药。
据世界卫生组织发布的《世界癌症报告2015》指出,2015年全球肝癌新发85.4万人,死亡81万;其中中国新发46.6万,死亡42.2万,严重威胁全人类的健康和生命。由于大多数肝癌患者具有基础肝病,起病隐袭,早期症状不明显或不典型,早诊比较困难,确诊时往往丧失了手术时机,介入和消融等局部治疗有效,但是常会发生转移进展,因此,对于中晚期肝癌,系统治疗必不可少。
国家肝癌专家组组长、中国临床肿瘤学会肝癌专委会主委和解放军南京八一医院全军肿瘤中心秦叔逵表示,“分子靶向药物索拉非尼一线治疗可以明显延长中晚期肝癌患者的生存期,成为标准治疗;可是用药后部分患者还会发生疾病进展,急需二线治疗药物。瑞戈非尼作为更新换代的靶向新药,在欧美已获批肝癌二线治疗适应症,本次在中国经 CFDA 快速优先批准,可以解决该类患者人群迫切的治疗需求,值得在临床上推广应用。”
据了解,作为一款口服多激酶抑制剂,瑞戈非尼可阻断涉及肿瘤生长和进展过程中涉及血管生成、肿瘤形成、转移和肿瘤免疫的多种蛋白激酶,包括 VEGFR 1~3、TIE-2、RAF-1、KIT、RET、RAF-1、BRAF、PDGFR、FGFR 以及 CSF1R。这些激酶单独或共同作用,控制肿瘤的生长、间质微环境的形成和疾病进展。
2016年12月发表于柳叶刀(The Lancet)杂志的国际多中心Ⅲ期 RESORCE 研究评估了瑞戈非尼用于索拉非尼治疗后出现进展的肝癌患者的疗效和安全性。其结果提示,瑞戈非尼显着改善应用患者的总生存期(中位 OS 10.6 个月 vs. 7.8 个月,P<0.0001),与安慰剂对照组相比,瑞戈非尼组患者死亡风险显着降低 37%(图 1)。
图1 瑞戈非尼显着改善索拉非尼治疗后进展肝癌患者的 OS
无进展生存期(PFS)方面瑞戈非尼组同样具有显着优势(中位 PFS 3.1 个月 vs. 1.5 个月,P<0.0001,图 2),以上治疗获益在所有预先界定的亚组中均得以维持。此外,经瑞戈非尼治疗的患者还具有更高的缓解率和疾病控制率。
图2 瑞戈非尼显着改善索拉非尼治疗后进展肝癌患者的 PFS
RESORCE 研究的探索性分析显示,从既往索拉非尼治疗开始,索拉非尼和瑞戈非尼序贯治疗方案可使肝癌患者中位生存时间延长至 26 个月。这对于苦无实质进展已久的肝癌内科治疗无疑具有突破性意义。
5、中国19个一类新药哪家强?上篇
在人人都谈创新的当下,中国药企除了在研发能力上构建自己的实力之外,还需考量的是与之相匹配的其它能力。我们就2008年以来获批的19个一类创新产品进行盘点(分为上、下两篇。下篇将于明日推出,敬请期待!),看创新企业除了打造产品之外,还需要构建什么能力?
12月11日,海通证券发布《浪潮之巅——创新药中美估值体系探讨》的研究报告,该报告表示,预计医药投资最重要、弹性最大的主线将于2018年正式启动。对于未来,其提出了4个判断:
第一,创新适量爆发。预计将有15~20个自主新药将在2018~2020年期间以最低每年4~5个的速度持续密集获批,产业的**将不断刺激和强化资本市场对创新的认知;
第二,创新质量提升。2020年左右,将有1~3个自主创新药在美国获批上市,国内创新药产业进入新阶段;
第三,创新是长期趋势。2013年以来不断增长新药临床申请将保障创新药产业的长期趋势;
第四,国内制药产业核心将从仿制向创新逐渐过渡。
从海通证券的分析来看,中国制药业的创新已经开始步入收获期,无论是本土市场还是国际市场,接下来的两三年可称之为中国药业崛起为的关键节点,由此开启的是一个新时代。
当然,在人人都谈创新的当下,中国药企除了在研发能力上构建自己的实力之外,还需考量的是与之相匹配的其它能力。在此前的一次会议上,十一届全国人大常委会副委员长、中国工程院院士桑国卫曾对“近年来获批的19个1类创新药产业化”做了分析。在此基础上,我们就2008年以来获批的19个一类创新产品进行盘点(分为上、下两篇。下篇将于明日推出,敬请期待!),看创新企业除了打造产品之外,还需要构建什么能力?
来自:桑国卫院士演讲ppt
1苏灵:单组份抢市场
国家一类止血新药“苏灵(尖吻蝮蛇血凝酶)”是利用我国特有的蛇种--尖吻蝮蛇体内的毒液研制出的新一代临床止血药。与国内已上市的其他多组分蛇毒血凝酶制剂不同,“苏灵”是高纯度、单一组分蛇毒凝血酶抑制剂,是目前国内唯一完成氨基酸全序列和蛋白质三维空间结构测定的蛇毒血凝酶抑制剂,作用靶点清楚,不含凝血酶原激活物,不激活XIII因子,只作用于纤维蛋白原。
国际上对蛇毒血凝酶的研究可追溯到1936年,我国研究人员于1998年发现毒液中存在有止血作用的活性组分,10年磨剑苏灵,该产品上市后便成为国内止血药市场的代表产品,被医院多个科室广泛用于减少手术出血。及控制术后、创伤、疾病引起的出血,成为血凝酶类止血药细分领域唯一的国家一类新药,上市3年后销售额即跃居国内血凝酶制剂市场第一位,并连续3年占据终端市场销售额第一位。2015年实现销售4.36亿元。
由于适应症广泛,苏灵已经成功实现全国各省区市的全面覆盖。南方医药经济研究所数据显示,苏灵在医院市场的终端销售价格保持基本稳定,产品竞争力较强,在全国各省、自治区、直辖市超过2100家医院实现销售。
2优朋:发力地方医保
盐酸安妥沙星(优朋)是由中科院上海药物所科学家自主研发的第一个具有自主知识产权的国家一类氟喹诺酮类抗菌新药,也是中科院实施知识创新工程和国家实施“重大新药创制”重大科技专项的成果。
1997年,上海药物研究所申请了盐酸安妥沙星及其系列化合物的专利,2000年获中国专利局授权的化合物、合成工艺、抗菌药用途等专利权,2001年安徽环球药业股份有限公司接受安妥沙星专利转让。该药于2009年4月15日获得CFDA颁发新药**。
进入医保目录是新药放量的关键性因素。米内网中国医保目录大全数据库显示,安妥沙星进入了包括广东、湖南等10个城市的医保增补目录的乙类药品中。2014年安妥沙星在我国城市公立医院化学药全身用抗细菌药市场中的销售额超过了1000万元,比2013年增长了30%。
3优诺安:主打安全性
与传统的硝基咪唑类药物相比,优诺安最大的优势表现在显著减少神经系统不良反应,安全性良好。且针对对神经系统毒性抵御力低下的老年人及儿童,优诺安的用药安全性尤为突出,这一特点助其成为相关领域老年人及儿童用药的首选。
2014~2016年以来,优诺安的销量增长速度分别为12.56%、19.5%、18.18%。
4丽康乐:增长快速
当前,我国生产爆款药“神经生长因子”注射用鼠神经因子的厂家共有4家,分别是未名医药的恩经复、舒泰神的苏肽生、武汉海特的金路捷、丽珠医药的丽康乐。苏肽生、恩经复 、丽康乐这3个鼠神经生长因子的2016年销售额合计达25.3亿元,与2011年底的3.4亿元相比,市场规模在5年时间里扩张了近8倍,年均复合增长率高达49.4%,堪称中国神话。
鼠神经生长因子的适应症主要是促进神经损伤恢复,用于治疗视神经损伤,以及正己烷重度性周围神经病。作为一种神经滋养类药物,鼠神经生长因子在临床上的应用极为广泛,是很多医药科室如眼科、神经内科、神经外科、骨科的常用药物。
虽然目前仍在4家公司中占有最小的市场份额,但借助自身集团化营销优势,丽康乐成为4家竞品中成长最快速的产品之一,市场份额逐年扩大,2016年为13.7%。2014~2016年,丽康乐销售额分别为1.8亿元、3.0亿元、4.0亿元。
不过,注射用鼠神经生长因子被多地列入辅助用药目录,随着目前市场限制辅助用药的趋势愈演愈烈,丽康乐的销售也会受到一定影响。
5津优力:国内首个长效G-CSF
粒细胞集落刺激因子(G-CSF)是国内外临床指南首推的放化疗相关中性粒细胞减少症治疗药物,包括短效和长效两种类型。Amgen在1991年2月20日推出了全球首个重组人粒细胞集落刺激因子Neupogen(非格司亭),迅速被临床广泛接受,近20年销售额一直稳定在10亿美元以上;2002年1月31日,Amgen又推出了全球首个长效G-CSF药物Neulasta(聚乙二醇非格司亭),第2年便成为重磅炸弹,市场份额一路攀升,2015年达到47.15亿美元的峰值,并且在全球几乎垄断了长效G-CSF的销售
国内有近20家企业研发上市了短效G-CSF药物,但长效G-CSF药物则仅有石药旗下百克生物的津优力和齐鲁制药的新瑞白,有逐渐取代短效G-CSF药物的趋势。石药集团百克(济南)生物制药有限公司的长效重组人粒细胞集落刺激因子(rhG-CSF)注射液(津优力)是国内首个长效rhG-CSF,2011年于国内上市。2012年在样本医院的销售额为20万元,2014年便达到1257万元,增长60余倍。
6益可宁:全球首支戊肝疫苗
戊型肝炎是一种经粪口传播的病毒性疾病,在卫生条件恶劣的低收入国家常有较大规模的季节性爆发。世界卫生组织估计,全世界每年大约有3400000例急性戊肝的典型病例,70000例死亡和3000例孕妇死胎病例。
1998年,厦大国家传染病诊断试剂与疫苗工程技术研究中心就启动了对戊型肝炎领域的研究。2007年,疫苗在江苏完成III期临床试验,2010年8月,国际著名医学期刊《柳叶刀》刊发了这一临床试验结果,标志着我国在戊肝疫苗研制上的世界领先地位已赢得国际权威认可。
2012年底开始,益可宁开始申请WHO预认证相关事宜。2016年9月,世界上唯一一个致力于发展和引进改良疫苗及创新性疫苗以保护世界上最贫困的人群的国际疫苗研究所和厦门万泰启动了关于为全球供应世界首支戊型肝炎疫苗益可宁的合作项目,这意味着益可宁走向国际化的第一步。
7普佑克:药价谈判实现放量
普佑克是我国“十一五”重大新药创制科技重大专项中的第一个治疗用生物制品一类新药,用于治疗由血栓引起的急性心肌梗死。普佑克是通过基因工程方法制备,具有特异性溶栓、快速溶栓和出血副作用低的特点,各项指标均达到或优于国外同类产品。
此外,普佑克的研发、产业化乃至上市,也在我国建立产业规模的哺乳动物细胞连续灌流生产体系,标志着我国在这一领域进入世界先进水平。
2017年,普佑克通过药价谈判进入国家医保,降价幅度仅为17%,上半年普佑克销售额为3730万元,同比增长167.2%,全年销售额预计在一个亿左右。由于国内心血管疾病药物市场广大,国内心血管患者约2.9亿,其中每年约有250万例心肌梗死发生,普佑克凭借其良好的溶栓效果,成为该领域的重磅品种,东兴证券预测,进入医保后,普佑克将会逐渐放量,未来3~5年有望成为十亿级别的品种。
8恒扬:火箭速度
艾瑞昔布(恒扬)于2011年获批,剂型为片剂,全球首创。该药上市后成为辉瑞塞来昔布的主要竞争对手,在缓解骨关节炎患者疼痛症状的效果和胃肠道安全性方面与塞来昔布相当,心血管、肾功能安全性更优,依从性好。
据样本医院数据统计,2012年艾瑞昔布用药金额达103万元,2014年为699万元,2015年为1095万元,同比增长56.7%,增速较快,预计2016年销售额为2117万元,较同比增长93.3%。2013~2016年销售增速分别为105.1%、56.7%、93.3%,连续三年高速增长。
随着人口老龄化的推进,慢性病发病率逐年增长,骨关节类药物市场是一个庞大的市场群。艾瑞昔布是国内首家昔布类自主品牌,恒瑞凭借产品自身优势及庞大的营销网络,再加上新版医保目录的推进,未来几年产品销售将会逐渐放量。
9艾坦:营销网络强大
恒瑞医药艾坦(通用名:阿帕替尼)于2014年10月被CFDA批准在中国上市,公开资料称为其是全球第一个在晚期胃癌被证实安全有效的小分子抗血管生成靶向药物。
替尼即酪氨酸激酶抑制剂,其具有阻断一种或多种蛋白激酶的作用。2002年,诺华开发出的世界上第一个替尼类药——格列卫(甲磺酸伊马替尼)登陆中国,甫一上市便成为治疗慢性粒细胞白血病的特效药物。由于其抗癌光谱、高靶向性、疗效显著等临床优势,带动了整个替尼类药物逐渐成为各种肿瘤治疗一线用药。之后的10年间,国外有16种替尼类药物接连获批。
艾坦作为恒瑞医药首个替尼类品种,无论是业界还是恒瑞,都对其充满期待。艾坦上市之后,恒瑞以强大的学术与营销资源向其倾斜,上市第一年销售额便达到3亿元。2016年更是呈现爆发是增长,达到近10亿元的销售,重磅之品的样子日渐清晰。2017年则通过国家药价谈判进入医保,由此奠定了其进一步快速放量增长的基础。不少分析认为,随着其新适应的不断拓宽,未来有望成为30亿元销售规模的大品种。
艾坦的成功,一方面得益于其产品力,另一方面得益于恒瑞在肿瘤领域数十年构建的强大销售能力及网络,能让产品甫一上市便实现迅速的放量。
10爱普莎:多势能
2015年5月,爱普莎(西达本胺)在众多光环加持下获准在中国上市。外界报道称,其填补了中国T细胞淋巴瘤治疗药物的空白,也标志着我国基于结构的分子设计、靶点研究、安全评价、临床开发到实现产业化全过程的整合核心技术与能力得以显著提升,是我国医药行业的历史性突破。此外,还有《时代周刊》、《福布斯》等杂志对其报道,在整个产业界造出了创新药上市轰动的大势。
从产品力的角度而言,近十年来,科学家们发现表观遗传在肿瘤的发生发展中扮演了十分重要的角色。西达本胺作为选择性组蛋白去乙酰化酶(HDAC)抑制剂类表观遗传调控剂,也越来越深入地被了解到,能够激活抗肿瘤细胞免疫,通过表观遗传调控基因表达重编程效应而非细胞毒作用带来临床获益。因此,联合西达本胺这种独特的表观遗传药物与其他药物的综合治疗方案来缓解或解决治疗中出现的耐药、转移及复发,和增强肿瘤抗原的诱导表达以及激活抗肿瘤细胞免疫,是未来西达本胺的重要方向。
对于产品上市之后的销售情况,从公开资料而言,并无公开数据。值得一提的是其通过药价谈判进入了医保,这对其接下来放量颇有益处。对于该产品而言,其最大的品牌背书是有多光环,是其开拓市场的加速器,但最终市场竞争依然要仰仗地面团队的运营,这是其当前最需练就的功夫。
6、昆药集团双氢青蒿素片进入Ⅱ期临床试验
14日,昆药集团股份有限公司(简称“昆药集团”)发布公告显示,近日,获得中国医学科学院北京协和医院《药物临床试验伦理委员会快速审查批件》,对公司申办的“一项多中心、随机、双盲、安慰剂平行对照、叠加设计的临床试验,初步探索双氢青蒿素片对系统性红斑狼疮患者的有效性、PK 特征及安全性”II 期临床研究方案审批同意,标志着公司的双氢青蒿素片研发项目将准备招募患者进入临床Ⅱ期试验。
项目基本情况:
药品名称:双氢青蒿素片
剂型:制剂
规格:20mg/片
包装规格:30 粒/瓶
治疗领域:红斑狼疮进展
阶段:临床 II 期
据了解,该新药研发项目是昆药集团于2016年8月以里程碑付款的合作形式,以7000万元的总价向中国中医科学院中药研究所购买的由诺贝尔奖获得者屠呦呦教授团队开发的双氢青蒿素片新适应症-红斑狼疮研发项目。截至2017年11月,昆药集团对该研发项目已投入研发费用约2299.69万元。
根据中华医学会风湿病学分会发布的《系统性红斑狼疮诊断及治疗指南》(2010 版)显示,“我国大样本的一次性调查(>3 万人)显示国内SLE的患病率为 70/10万人,女性则高达113/10万人”。SLE发病机制复杂,随着疾病发展,患者往往出现多系统受损,严重者常危及生命。目前临床尚无根治SLE的方法,抗疟疾药物、非甾体类抗炎药、激素、以及生物制剂是主要获批的治疗用药。
根据IMS纳入美、德、英、法、意、澳、日、韩等主要医药市场37国(未包括中国)的销售统计数据显示,2015年相关国家的SLE医院用药市场达9.36亿美元,这些药物大部分为针对SLE无药可根治的局面下进行的对症治疗,即便如此,2011-2015年这一市场的5年复合增长率仍达到17%,保持了稳定良好的增长。作为一线用药,抗疟药是现有SLE市场的主力军,据GlobalData数据统计,2012年抗疟药全球处方量在整体市场中占到了35%,同时预计该份额将进一步扩大至2022年的38%,并继续保持其领头羊地位。
昆药集团方面表示,红斑狼疮的治疗药物是目前未被满足的临床需求之一,具有广阔的市场开发价值,该新药研发项目符合公司专注慢病领域的战略定位,项目的实施可进一步发挥公司在青蒿素产业链上积累的优势,提升资产收益率,拓展发展空间。

| 欢迎光临 药群论坛 (http://www.yaoqun.net/) | Powered by Discuz! X3.2 |