药群论坛
标题: 【FDA】非无菌半固体制剂扩大规模和上市后变更:体外释放试验和体内生物等效性要求 [打印本页]
作者: xiaoxiao 时间: 2021-8-16 02:12 PM
标题: 【FDA】非无菌半固体制剂扩大规模和上市后变更:体外释放试验和体内生物等效性要求
本帖最后由 xiaoxiao 于 2021-8-16 02:14 PM 编辑
& T1 D0 M W- C( K
1 B# E' O& W4 f' t- `- j. ?7 c【FDA】非无菌半固体制剂扩大规模和上市后变更:体外释放试验和体内生物等效性要求
/ M) P) a t1 d; ~8 G/ W6 p/ k! B" B/ b' w" [% u& }; V) s+ p5 f
/ S6 Z4 o4 X' X' j
非无菌半固体制剂扩大规模和上市后变更:
J3 e) o3 P5 T8 e 3 ?& h% @8 O. K0 q) }
体外释放试验和体内生物等效性要求
Z6 Q5 {' H6 b7 f2 ^. j
3 p$ v! ?9 B- y m3 j" v/ ~SUPAC-SS: N**terile Semisolid Dosage Forms; Scale-Up and1 z; U0 L& U+ ~! V; ?. W
" U( u2 A0 F1 Y3 bPost-Approval Changes: Chemistry, Manufacturing and Controls;7 c: c" c+ k3 x* ~& l
- S! Z: p* c( w# P$ @6 o' L
In Vitro Release Testing and In Vivo Bioequivalence) B, R- U, j4 S* K% Z% e
' g. ]# i2 `) y: u( V/ T* D
Documentation
4 [3 |) J; D$ V
. u# r, i' C& V8 K! g; H3 b8 B
/ Y' `8 F0 |' U2 g7 M
1997年5月美国FDA发布2010年3月药审中心组织翻译费森尤斯卡比(中国)投资有限公司翻译药审中心最终核准
, b: n8 \& C3 C6 t 3 l6 [! s: z% R# d" M
; e* ]( _6 |: a/ ]目 录
2 ~5 W) K: `4 g/ s- l0 G, R
1 ?4 j. [) @9 ^I. 前言 3# I I, ] k. e% d9 s- ~# `* \
8 G3 C2 Z4 C( r) z- }$ aII. 背景 41 E" u5 j! l4 ], j6 s2 Y
% E# o' e! s* K6 ?A. 关键的工艺参数 4
* R2 w3 O6 @ U
$ j% Q: a Y; g& e; | r, n1 WB. 稳定性的一般考虑 4' J; u& n7 T( z5 Y6 a9 X" t
" F. L$ M5 M$ u* z6 ?- m
C. 体外释放试验 5- Y5 r% B" x4 J, d
' O8 c r1 n3 r
III. 成份和组成 53 k1 W$ M( c8 ~" b5 v
9 i# g2 o# B/ f1 O n
A. 1级变更 6
4 N. J. p1 g+ F- w' W, I( W
# K3 w z- V: B2 d. \: sB. 2级变更 6, K" s+ d* P- m- Q
# \4 t( B2 z9 a( D
C. 3级变更 7, e% _: ~1 @7 @7 I, d9 A2 d; O4 Z
, E0 ~, h/ ^4 X7 GD. 防腐剂 82 ^4 u6 c$ j9 S+ q9 M3 K
1 U/ Z/ v' D" X" ]IV. 工艺 9
$ T/ s! i- v" Z# q ( w6 w i. T7 r
A. 设备 9" Q( r' ?4 f! W* W! ~2 O# W
& U: a5 _% n( XB. 工艺………………………………………………………………………….11
! o* ]- @1 @' T( y- l 0 _7 z* s' w" z( h4 `7 J/ D3 q
V. 批量(扩大规模/缩小规模) 116 n+ O8 E M0 z
3 N# x" U( Y% q& J: z' o2 QA. 1级变更 127 g* t t4 L9 E) @8 S+ ]
& j0 W; ]: {$ C$ f
B. 2级变更 12
' r$ s1 n0 j' X $ n5 U. q( V$ |% Q! I- [
C. 3级变更 13
7 L; t: p3 R8 X' C5 r
+ X8 p$ x4 X% \VI. 产地 13
( c8 _) x: D# @8 n" S : F) P4 X* V& g* i$ G! y
A. 1级变更 13
+ R3 T6 }4 D: ]# d8 v 2 H1 ?6 y& _5 \. l
B. 2级变更 14
6 k' w4 Z, c m' Y& H0 H ; l$ L: e+ j1 {9 G
C. 3级变更 14; W+ H' n" Y9 t% `
) c ]4 J$ u9 [" {" W5 V5 ^/ [VII.体外释放试验 15+ j; E% F( H C. b+ H" |
* Z: a" k- d, k3 p6 l' F0 O( @0 j! K
VIII. 体内生物等效性研究 20- f8 a- [5 n1 v* |4 p( A" l3 }4 L
/ |# F, L- m8 `4 T+ I/ I术语解释……………………………………………………………………………..22
5 }6 B9 |! k5 `
' Q5 f5 @1 O2 M8 S+ ~3 t. b$ N ( G; J3 l+ M* M) L* |* a; ]$ v, `6 h8 n
2* \# i) K5 g e1 q
" P3 e0 k C9 w- U, K非无菌半固体制剂扩大规模和上市后变更:体外释放试验和体内生物等效性要求
5 y1 c' t* Q# O
4 r& |( o8 q; B5 ZI. 前言
5 ~/ a, F( G0 [6 z8 t/ \ [$ D1 ~
- s& L% \$ C0 S: I$ }本指导原则旨在向计划变更已上市半固体制剂的(1) 成份或组份;(2) 生产(工艺和设备);(3) 扩大/缩小生产规模;和/或(4)生产地的新药申请(NDAs)、简化新药申请(ANDAs)、简化抗生素药物申请(AADAs)的药品申请人提供建议。本指导原则涉及的是局部用药的非无菌半固体制剂(如乳膏、凝胶、洗剂和软膏),并就(1) 变更的级别;(2)建议的化学、生产和质控(CMC)试验,用于支持各级别变更;(3)建议的体外释放试验以及/或者体内生物等效性试验,用于支持各变更级别;以及(4) 用于支持变更的资料,进行了定义和解释。; m; p- y+ v$ T& f3 ^, Q
8 ]$ e: H( `6 t, N4 j( n本指导原则详细说明了应提供给药品评价和研究中心(CDER)的申请资料,以确保特定变更的半固体局部用药制剂的产品质量和特性保持不变。本指导原则未评论或以其他方式影响CDER执法办公室(Office of Compliance in CDER)或FDA药政事务办公室(Office of Regulatory Affairs at FDA)确定的执法/监督文件。9 \8 n3 b) T: g
. [9 A: N3 D9 M0 }; o3 i1 J本指导原则就下列多种变更的试验和归档文件提供建议,提交适合的试验和申请资料:(1) 多个1级变更(附有1级试验和备案资料);(2) 多个1级变更;1个2级变更(附有2级试验和备案资料);(3) 多个2级变更(附有2级试验资料和1个Prior approval supplement(PAS));以及(4) 3级产地变更以及任何其他1级变更(附有3级产地变更试验和申请资料)。用于支持变更的资料取决于半固体剂型的类型和复杂性。对于Changes Being Effected(CBE)的补充申请(21 CFR2 k7 M0 _' Q/ i& F6 y( z5 D7 j
- F" x. \, h7 V8 c* V0 }3 14.70(c)的变更,FDA可能对补充信息进行审评,并决定变更不批准。申办者应与相关的CDER审查部门或人员联系,以获取本指导原则未涉及变更类型或短期内连续的2级或3级变更的实验和申请资料的信息。
* q5 |7 Z1 ]. M) l5 d/ d4 T & D# s G% H: i6 b0 R' m& H
法规指出,申请人可以按照联邦公报公布的指导原则、通告或法规对某一已被批准的申请做出变更,以减轻繁重的变更通告量(如,在提交补充申请或在下一个年报中通知)(21 CFR 3 14.70(a))。本指导原则允许在§ 3 14.70(a)
5 C: c' M6 t; w( S. M) O9 M ! O/ ~# Q1 _2 o1 \* v9 x/ u
0 x) R7 j s% q$ b5 K3 i: S X' S5 I
31 {( p0 {6 F( Y& `
% j2 Z& h, h+ v9 b4 I
范围内减少某种上市后变更的通告。
/ p+ ?- h4 O+ w* h 7 \" g1 H8 F% s! z$ B0 k. V3 b
AI. 背景
: C! Z4 e3 Q0 `8 h5 J) t4 z , k3 h7 \+ C* T1 J) ]0 `
通常,半固体剂型含有复杂结构的复合处方,常包括两相(油相和水相),
$ Y7 j- c# x7 d+ m( B ' j$ }" m: g/ k$ E( a% P
其中一个为连续相(外相),另一个为分散相(内相)。活性成份通常溶解于一7 A; P2 }3 \ ]6 ?6 `, e' C% o
4 e: P4 N2 n( d/ g' Z& e9 J2 b6 K
相中,虽然有时药物在系统中不完全溶解,而分散于一相或两相中,因此形成一8 @" m; v( V- [# R. Z5 p( U+ b. z
3 G( @6 \" X& }5 Q7 S1 E' X
个三相系统。该剂型的物理特性取决于多种因素,包括分散相粒径、各相之间的
9 R" I5 P/ ~( c5 u" ~( W% L 7 U* X$ l$ V5 }0 f
界面张力、各相之间的活性成份分配系数,以及制剂的流变学性质。上述因素共1 I2 a' y% V% i: b6 X
6 x6 P% R* L9 [* T- P4 f. W同决定了药物的释放性能,以及其它特性(如黏度)。9 q" F* E' U" T- \& o' j6 [
$ |) `* Z9 L# Y N
A. 关键的工艺参数7 p) q# x1 g/ ~+ H9 v' @. b
$ b/ o `; C+ Q% g, B, ]" L
对于真溶液,溶质加入溶液的顺序并不重要。但这不适用于有分散相的处方,
+ V6 R; R* o- f1 u 0 c2 e6 u ]# i2 k1 a
因为分散物质的分布可能因其加入相的不同而存在差异。在一个典型的生产过程
9 O. p6 V, i/ Z$ }( ?" W * ^; J( D9 S8 A6 L6 R" {
中,关键点通常是指一相系统开始分离为二相系统,以及加入活性成份的这个关' O- }4 A2 \- a# Q9 \5 q$ D
& s3 Y( | k, M键点。各添加成份的溶解度是非常重要的,因为它决定了混合物的外观是否为单- V8 x% R* o9 L
; z( [* J j- A H一均匀分散体系,这些数据应尽可能由光学显微镜检测的溶解度数据,以供审评。& M0 g& T3 B1 h( S
4 p; q8 i7 S6 g! R/ l
溶解度数据对于将溶质加入处方至接近或超过其溶解度(在任何温度下)的浓度,
# {9 F' j2 _* x0 M( Q* v: K
3 Y V8 T9 V q" P而制成成品尤为重要。
$ F! t$ i& V4 ? 8 @1 y/ B4 f- |4 U
在生产过程中的上述任何一个时间点后发生的偏差,可能是影响成品特性的
* {" J! C) X9 o. v 0 q% ]/ x6 Y5 p
关键因素,尤其是在企图通过减小液滴或粒径增加分散度的工艺(如均一化作
9 M7 K, ?0 Z% Q# U. ~1 U
! }& p5 ^3 c1 j. M1 M3 ?5 H' x用)。成品在包装前的老化至关重要,应在工艺验证研究予以重点说明。: Q+ M& b8 T3 A9 i
; L( d, u: ~) ]8 OB. 稳定性的一般考虑
% H3 J! ]6 \' G& B$ @1 b1 S9 Y % [. X9 Q4 o* Y( j, R5 F8 ^7 ?
应评估扩大规模和上市后变更(SUPAC)可能对药品稳定性的影响。进行- a; m, Y! J0 d8 s. _1 e
( f1 q6 t3 x* |5 y4 z# p& F1 {稳定性研究的一般指导原则,可参考FDA的人用药品和生物制品稳定性申报资料8 v( L* E% I6 W7 P7 X
( O3 a( W4 l5 k( @7 _1 T8 M' A0 \3 ^指导原则。对于SUPAC申请,应该考虑以下几点:9 S, V& K( m( |4 Q9 }8 I+ I
8 q- V6 x+ ?: F1、在大多数情况下,除涉及扩大生产规模外,可以接受中试规模批次的稳: c" n. \2 {8 r, F
% z6 ^8 O; P9 x' M5 M+ e- Z( A9 l
定性数据用于支持变更申请。5 N* k0 V5 D; ?: H
/ P) q2 }6 k8 h3 S) o% D/ ?. F9 H
2、当稳定性数据表现出在加速条件下效能降低或降解增加的趋势时,建议
3 X. g" f" v% y- ~$ Y
+ _. C) g* t6 i m8 d1 n提交变更前有代表性批次的加速稳定性数据,便于比较。在上述情况下,还建议
& r9 s6 W) H y& @# k # {2 X1 w* n0 ^% R1 J: l
在补充申请中应提供全部正在进行的试验批长期稳定性试验数据。提交既往加速+ L4 d/ j0 A# S8 p& Q
: q& g; Z9 y4 V9 H) K9 i/ s稳定性试验数据和已获得的长期稳定性试验数据将有助于补充申请审评和批准。* _" m: q1 t# d
3 N. |3 `5 X/ p+ R+ o! v7 z, k/ [' E$ J
3、稳定性承诺包括在有效期内,按照已批准试验方案,对首批或前3批(详
! w E; y* o0 L* j/ x3 m* Y/ T2 ~
7 h: F8 x8 { r( D" o! U
- K; ]! S& z& ~' _# z' N! t4
2 b8 [2 q9 {# b% m7 z1 I
* P& @# m: ~2 Z3 S见第III-VI节)产品进行长期稳定性试验,并在随后的年度报告中报告试验结果。
0 D( p0 h2 t8 q! B: ` * c X- q4 @3 n
C. 体外释放试验
# B* i& E/ s2 M, D) c) a1 K
8 ^' g; ?* r- j9 t. r- M% \; V在对照的临床试验中证明其有效性是所有药品的重要依据。鉴于此类试验相关的时间和费用,不适合作为常规质量控制方法。因此,体外替代试验常用于确保产品质量和特性不因时间和这种变更而改变。对半固体制剂及其组份的各种物理和化学检测(如活性成份的溶解度、粒径和晶型,制剂的黏度和均一性)已提供了产品性能一致的合理证据。最近,体外释放试验已显示能全面确证活性成份从半固体制剂中的释放行为。$ L/ F. }6 y' ^8 X
, y( @2 v5 j; t9 _( ]7 r% c) q I- @* e7 N: B
体外释放速率是一些理化性质的综合体现,包括活性成份的溶解度和粒径以及剂型的流变性。在大多数情况下,体外释放速率有助于评估制剂在变更前后的同一性。然而,也有该试验不适合用于上述情况。在此情况下,,应提出检测制剂同一性的其它物理和化学试验,并与审评机构进行讨论。在所有试验中,均应考虑质量特性“等同性”文件中的计算和统计方法。
& b* l6 K+ L4 g ! j1 K$ R; r. O/ L5 o3 a
此时获得的半固体制剂体内外相关证据,不如口服固体制剂体外溶出试验替代体内生物利用度令人信服。因此,药品审评和研究中心目前关于体外释放试验的意见如下:2 g0 \; B" ?+ H, g7 N
\$ d4 h0 L3 M! E; \3 D- B1、对于半固体制剂的某些扩大规模和上市后变更,体外释放试验可用于评
# c0 F" T+ m% \9 l( ] & Q7 c7 Q" Z; ?& [
估产品的“等同性”。$ U+ N1 X* F5 b, L0 t
$ Z* \- W& D: K: t2、批准NDA、ANDA和AADA时不需要研究和验证体外释放试验,也不要
" b. f5 E% }. h& R) |3 Z( z 2 @% y* T% [" n
求将体外释放试验作为常规的批间质控试验。
O$ E) i; v' {* P% ~8 a6 Q: ~
- n Y9 }' X( i3、不能仅用体外释放试验替代体内生物利用度或生物等效性试验。8 J( S4 @& V7 \0 Q9 A% i- E
+ w, o5 B3 R0 s& V
4、不应将体外释放试验用于比较来自不同生产商的制剂。
; X( d' c! ]$ v. X* j
8 q# ^/ [2 G4 _% P, w7 r0 lBI. 成份和组成8 V) H* l0 i) T; M/ n; }2 K
- G7 Q7 o* v; r) F- {6 `
本节的重点是药品中辅料的变更。辅料的性质变更应该仅包括已获得批准的
% _) G; e. \' E( X
4 e) h5 N5 k! S$ \1 V用于特定给药途径的药品中已有的辅料。辅料数量的变化不应超出已经获得批准: m: q$ M0 V+ {6 v$ o+ N
0 ^2 m6 A3 E1 j# `8 {
的、给药途径相同的已批准产品中的数量1。应该提供成份和组份变更的年表。除
1 p0 d+ r3 M0 g# L i, s2 s T# E
9 U3 z, _& P3 _% v/ V/ D下文所描述的内容外,增加一种新辅料或减少一种已有的辅料的成份和组份变更3 M6 v( J" s. F8 D! J7 Q% a
4 z9 c: W- p e7 G( u" O! e1 k
定义为3级变更(见下文第III.C节)。上述变更通常需要更改标签。4 F7 K- X1 i- D/ _
file:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps16.png
- v- A5 O) T7 A: ~1 C 1 y4 [( m0 U* Y& a' T4 a
( i; f" L& v/ c2 B( [& _* y; K4 d/ Z
1 FDA, CDER, 非成份活性成份指导原则,1996, Division of Drug Information Resources.0 a" p& W% S8 T! F# n
& [- |8 P. d$ f* s' s1 D
$ E2 W: ~) b: O6 ]
5% {8 V) q% \5 ?) z( D' C
9 K, m! q( J! i/ M6 N, c6 a* T防腐剂的组份变更将被单独考虑,未将其列为第IIIA、B和C节中总累加效应的一部分。4 L- L4 Q7 r5 O# b' B
+ {8 b) O4 m! m$ U" g; A! b2 y! ~- G6 hA. 1级变更1 O/ J+ V, s2 T. l6 }: M7 p7 E
: N/ V1 a( b N) }, c ^$ G0 j* }
1. 级别的定义
) V6 g/ I( l1 Z- `5 s # p! W" s8 V+ n @# I# U4 ]
1级变更是指对制剂质量和特性基本不产生影响。
+ J* ^# w# s7 C; E0 b" ~
' b; n1 u4 u: R3 @+ q8 \4 m例如:
. B8 N7 R4 @' L/ d% y ! X( D# t& g4 h) |
z 去除或减少预计影响药品色泽、气味或味道的成份。) a% m% R! c$ o# f
( J' _3 m. ~% ?! {1 Bz 已批准的任何辅料量达到5%的变更。所有辅料变更的累加总和不应超过
7 a7 u5 e) Q( s
# ]6 U9 m7 E V5%。组份的变更应基于已获批准的基础上,而不是基于之前的1级变更。由辅料
" `1 G# ~. |3 s. P7 p& `% S
4 J& e/ d( M% B. D; y成份和组份变更可能引起稀释剂的改变(q.s.辅料),不受最多变更5%的限制。
6 i! }) e. N, L+ ]
- s; ]" x: r* A1 p/ A' B" b. ez 结构主要是单一化学实体(纯度≥95%)的赋形剂供应商的变更,或者其它辅料供应商或技术等级的变更。+ l. L$ U" A' {9 J% A' A, a2 @
, b+ o5 a8 @2 x i
1. 试验文件
% u- H1 C4 p1 u6 q
- b2 Q' N1 _8 C- @( v- F$ [! la. 化学文件
* L$ |4 K- _4 L* ^3 D1 s3 i" ~
( J# n! V3 C, U$ c0 \, ~3 U. J5 w+ A0 G产品出厂要求和稳定性试验。
t E! t- J) O2 { e6 A4 ? ( t6 |2 z5 P2 f
稳定性试验:在年度报告中报告的首个产品批次的长期稳定性试验。9 Q! F; J# M/ M7 M& C `' _% Y2 p: k
# t1 B& b# g/ z t7 o; k, z
b. 体外释放文件# ]6 J y: J* m
6 m+ h7 U: Z: h8 y无。$ x0 E/ }+ {4 P9 k
3 ` C0 p9 f- n! |7 E* Y/ o2 [2 Ec. 体内生物等效性文件无。
+ s* \/ N* P7 j- H$ h! V+ J8 g 9 m) Y3 O! s$ Q0 R
3. 备案文件
$ `# ]( D" R$ L2 Q* w9 l; P
: r9 [/ W4 x8 M+ ^( e年度报告(包括长期稳定性数据在内的所有信息)+ i \% ~5 U/ ~% w: J! a
& o: V4 C, U8 l* g/ sB. 2级变更4 f$ @& T' N8 w# O" h& C) x
/ _. e& U- F. W, h
1. 2级变更的定义
' [1 ~8 A' q \( ~2 o" D* b ) q% m2 Q/ c) b% E8 T/ {: ?
2级变更是指可能显著影响制剂质量和特性的变更。! E$ Z/ y) j7 P2 C) n7 r- f* W$ P0 t
% R3 f3 @2 l. v例如
5 V5 W% U+ k! i$ T5 R( } 9 C( W7 h6 W$ i! R
z 已批准的单一辅料量大于5%和≤10%的变更。所有辅料变更的累加总和不应超过10%。组份的变更应基于已获批的基础上,而不是基于之前的1级或2级变更。因辅料成份和组份变更所致的稀释剂(q.s.辅料)改变是可接受的,并且不受最多变更10%的限制。
! c5 s: P! A* Z: v
. L/ l8 g1 [1 X
* \8 z% w |/ M& p& f h6
: k( o5 H1 [1 S/ |$ J/ {; z 9 I" G1 y- J; z! V& p* ~( E& S
z 1级变更中未涵盖的赋形剂供应商的变更。# f0 r1 K5 A P7 W! P
$ x: ?( E. a( ?2 k8 `
z 赋形剂技术等级的变更。8 V# y! \) U7 u$ _; V& B q
+ k6 M: c* Z: _. C
z 如果药品为混悬液,原料药粒径分布的变更。# z5 H4 F1 N( N G
' d. j7 L' h! t0 Z/ ?7 q
1. 试验文件0 R& W N- M. t( g' Z5 U
0 j: {% w, L1 J0 |a. 化学文件
! h. V# [, D7 p' ~: g& f- W
( }% x7 L: x9 e产品出厂要求和已完成的批记录。
. V5 \- N2 ?1 V
! {- G+ h- `/ t8 B& \: Z" n稳定性试验:在changes being effected补充申请中报告1批3个月加速稳定性数
# P( y$ e# |* b0 c8 P/ D7 b/ e7 { 8 h, G4 @% ~: Y/ h. i1 ?
据,在年度报告中报告首批次的长期稳定性数据。
; h- v7 F+ i# m; l # k( ]) P1 F' A
b . 体外释放文件
; d1 n+ i' H" _7 j5 ?
6 \( P; D) Z2 q% q" D) o" B" K新/变更的处方批的体外释放速率应与同期生产的变更前处方产品进行比较。2 z2 v* `& O4 @# Q0 \9 c
: {0 }2 Q8 g0 z1 @: v9 B应确认2种处方的中位体外释放速率(以斜率推算,见第VII节)在可接受的限度
4 F8 y1 e: e5 @7 D8 ^/ f% X; ~ ! [4 o& l5 S# A B0 j/ R& ^
范围内。试验过程详见第VII节(体外释放试验)中描述的试验程序。
$ I$ k3 ]2 o1 |+ M& }. g & s/ C' N1 J' i2 c6 G
c. 体内生物等效性文件无。
- ]4 V! a# t) U" q' r g( i- M 6 d) Q( }( q, V* W! K5 c
3. 存档文件
: |' @- U8 h v% t6 O5 b
6 j% r, p4 C% ]1 y6 k5 O8 b/ Wchanges being effected补充申请(所有信息,包括加速稳定性数据);年度报告(长期稳定性数据)。! z% X2 O1 K7 H% b6 }7 g; b
4 O: M' ?5 ^6 A( \C. 3级变更
( ]' l$ y- |! w- v: Y: A+ U
' ?/ r% V9 o4 X6 Q+ ^1. 3级变更的定义
, O% d* j# ?5 p9 P \2 H+ l 9 j4 E4 B. p' y$ l( ^, ] ^
3级变更是指很可能显著影响制剂质量和特性的变更。 I, _4 ~/ n0 N+ L i' i
8 Q: _* C8 B( d5 Y
例如:7 S) f/ C# U$ E" @
4 z/ r4 ]2 g3 n4 F* [1 D' ~z 所有超出2级变更范围的辅料质量和数量变更。
. ^2 O) S9 P) b' J
' f. B" K8 J' u$ s% g7 A9 hz 如果药品为混悬剂,原料药晶型的变更。
" B5 F4 U" T" D+ F( E* y/ `" M
9 L( F' J( a* c! m# A; I4 a+ P1. 试验文件
+ A5 E* E; R! `2 `2 A2 T; \2 \ - R0 b( P1 X# z6 K2 k
a. 化学文件
9 N; Q5 q+ I! x) P/ }7 m* ? * W& D. H! X5 |
产品出厂要求和完成的稳定性试验。已获得重要信息:在批Prior approval2 `" K! I! q: [
) Y8 s2 |; f$ W2 q. }: k
supplement中报告1批3个月加速稳定性数据,在年度报告中报告的前3个生产批次
' I0 |' w' b# D& G7 c" v5 s8 @9 L ) e+ Z6 i* |9 \# Z! j; e: c
的长期稳定性数据。+ d8 d4 f# ^5 { `3 u
2 e) \8 W; |1 f4 T3 [$ ~* ?
尚未获得重要信息:在Prior approval supplement中报告3批3个月加速稳定性' m! A! Q' W, c8 w
7 p* B( w/ f: Z& k数据,在年度报告中报告的前3个生产批次的长期稳定性数据。
2 p# F0 j9 w" g ' N% M0 X) P; Q/ @- C2 h
7 t1 J8 \! C5 M \" ?! v% @- B
7
$ t' M9 ]! P6 |/ d) l# r" B: @6 N 9 ? ?% Z; ~8 |. k- k" n2 p* H* |
b. 体外释放文件
% u# s" u# V6 M! k5 e
% s% S0 g% J- {" ^+ r应该确定新/变更处方制剂的体外释放速率作为一个参照。在该3级变更情况1 _8 ]2 j5 Y" o2 m
5 I$ e0 @1 l3 M; u. ?1 ~7 N下,不要求体外释放资料,但是鼓励申办者基于本指导原则,获取该数据,以便用
+ T6 @" f$ D/ u! g( ~2 |4 {
0 ?/ @0 P1 l4 B于后续的变更。
0 c6 w( L7 Q9 r4 x- F & D: |" O+ q) ?2 o8 ]
c. 体内生物等效性文件
! `7 w4 M7 r, A5 N: @& U% I! [. L- O & w" m5 l# g, R
最高规格的完整生物等效性研究,较低规格的体外释放/其它方法。5 z* E. p' C" V H
3 E$ ^6 @6 W5 H5 M$ |; s
3. 存档文件* [' d6 Q" A" T0 {
9 Y; I7 Z& M7 q a4 b8 V
Prior approval supplement资料(包括加速稳定性数据所有信息);年度报告(长期稳定性数据)。
; G; V3 {! J6 L# f. q
3 R2 J" j0 j5 S) n2 [, Y; w5 @7 h) oD. 防腐剂8 l- d8 [ X+ { A' Z
, Y8 h1 M' ? w2 |2 [对于半固体制剂,任何防腐剂的变更均可能影响产品质量。如果在处方中做3 ]8 i2 h! g( E9 T8 W+ ~3 C6 N
8 c& u$ j. N$ |# D# ~9 F
出任何质量或数量变更,应该进行附加试验。对于防腐剂变更,不需要体外释放) v8 t' ^+ R3 w2 t3 \. j, J/ k: {
! p& _2 p L$ i7 p9 t0 p
文件或体内生物等效性文件。5 M' q. ~7 E6 S9 i, a
1 h2 A- B: l6 M: r" Q. ?) ~1. 1级变更
: r. {9 `0 \6 y% Q) @, t# ^' K; R
+ D1 q/ B' k8 I5 z* d4 z) C- w' s# ?a. 1级变更的定义' F3 Q0 @: `4 x- F9 D% g
2 F( ~) p; e$ s) ^2 c- U/ |
变更量为已批准防腐剂量的10%或少于10%。8 g7 i3 {* u; y3 I; Q9 T
8 }% y5 I8 n* S+ k! f T! Z# zb. 试验文件
4 j7 Z! n) m0 _, o1 d & j+ e$ Z% K1 R
z 产品出厂要求。
. W& k" d% v" q# p; z) o E; t- i4 v2 \3 a2 l }
z 在最低的有效防腐剂水平进行防腐效果试验。 c. 存档文件
. q) X Z# U4 T# f" F1 C6 ^
4 W' o; R0 q3 v年度报告
: t" H+ ^: V3 {* |" R) a& o8 J/ S
, ^2 ]# r, p/ x+ P0 S2. 2级变更
/ i% [( g! C2 Q7 @( T& F 9 K0 @: o. E7 r" X+ }4 N0 B
a. 2级变更定义- T. ~$ n- \( Y6 X5 x
7 d! L3 \: t9 v. _/ u! Y) o6 V
变更量超过已批准防腐剂量的10%,最多20%。' F- Y7 v1 I9 _& u4 F2 b) j& W
* Z9 { U/ j0 ^ T5 L7 H& V8 ]; W; V
b. 试验文件
3 f1 m5 Q- e! p9 ^. X $ u L1 O+ @) V$ N. q* Z% `- f' e) t' B
z 产品出厂要求。4 w: |9 b/ {6 ?$ | R
3 X' R, X+ }' O' m, l5 W, J; N
z 在最低有效防腐剂水平进行防腐效果试验。 c. 存档文件. R( K5 T. `) X% B+ A4 H
k. g1 b" G1 S" T" C8 ~% lChanges being effected补充申请。9 A! Y$ t4 p/ n
' L$ y( l) n, k, W3. 3级变更* j" W0 j0 c3 X- O$ G. Z
3 F, `! g" ^1 V, c
" |+ c5 Q* w& `2 ?7 ~! `' D/ r8
. O9 ^* H6 G2 X" ^ + Z' f3 w T# Q; m$ G
a. 3级变更定义; X1 t2 p0 P* G: @7 v
4 y9 l4 l. d2 I; P- e" x2 B变更量超过已批准防腐剂量的20%(包括删除)或使用不同的防腐剂。# F$ x6 N8 j. {) P2 k
( D C* _ w; A5 J) V$ C8 E6 q' f
b. 试验文件
1 y6 [: m; |1 d2 u 0 m, q6 B# {6 N: g0 ~5 L
z 产品出厂要求。9 G3 [+ D0 Y9 ]
, b) {, ~* J; c( j' K* k4 F
z 在最低有效防腐剂水平进行防腐效果试验。
$ s6 p, ^* y4 g1 Z1 ~3 r/ ~
4 k8 m% M, d. Yz 新防腐剂的鉴别和含量测定方法。
* j0 h( x# }) j
4 c' p3 i# v! z gz 表明新防腐剂不干扰试验的验证研究。8 R8 g5 J6 t9 @7 u
! y3 D# T. a' p4 L
z 完成的批次记录。
/ u9 A2 S* U. T- B
7 l) w8 ~' T/ y& ^z 稳定性研究:在Prior approval supplement中报告1批3个月加速稳定性数据,以及在年度报告中报告首个产品批次的长期稳定性数据。
& m: A- Q. q$ Q, `( x
4 ~$ j: ]2 l+ Q+ J- `* `c. 存档文件. n, Y* k9 m+ [( s0 H. P$ p
3 `7 `/ E. _6 ^. q2 M' t H* M
Prior approval supplement(包括加速稳定性数据的所有信息);年度报告(长# p/ ~ L5 L+ @$ B
+ R% m. @$ }5 l6 R7 D* `+ {: Y1 v期稳定性数据)。
c, ~. \1 t% g
9 B' ^/ }0 m# u, w1 }2 k; l% t0 `IV. 工艺3 L$ e/ u0 x; ]7 P3 J0 w( T
8 A6 U+ T+ B% x: S) X9 j
工艺变更可能影响生产过程中所用的设备和工艺过程。+ @! R! X2 X# p" K
$ ~0 r7 ~4 _& ?+ g- B! [2 @+ UA. 设备% n9 P+ [2 a& |. q3 P
/ e- C: s. `. L% [1. 1级变更
3 l0 S7 I. H. }4 E O
2 L6 B8 ~4 o. E9 d; E1 M0 la. 1级变更的定义+ |+ k# a2 B+ l
+ N* J; a" n! [; F/ {; d+ A8 x
原料的转移设备由非自动化或非机械化变更为自动化或机械化,变更为相同设计和操作原理的设备。0 C' p& s2 D. G; ^
2 F# u0 u* {) wb. 试验文件
N& ]; V' h7 m; j 6 w, {6 S0 ~' x# a& F' y7 L! ]1 M
i. 化学文件3 |4 I0 R& X' j" ~5 h
' n8 B" G" `9 m( O
产品出厂要求。变更通告和提交更新的已完成的批记录。" T6 L: i' W3 Y; y! {
, _8 j; T# S- U H稳定性试验:在年度报告中报告首个产品批的长期稳定性数据。( T, D( t; h6 F, W+ K* n* V
0 K3 ]( ]" D- z/ O w; r- ^ⅱ.体外释放文件7 M# E0 E# c% g' O3 Y
( }3 S: O) }, @) v ]9 V5 F无。. G! r3 [ Z0 x) ~( t& D
2 C7 L' l2 e7 k; X( r0 v
iii.体内生物等效性文件9 X+ a+ F* P9 j4 A, p9 |( Z
" X5 s; \) K+ R$ B% @ X" U
无。$ t- ?0 E. ]& F7 J: @& T
. z6 u8 k$ e9 Pc. 存档文件( O+ [! z6 g& x, M/ E3 c0 y2 Z% {
! n- {* A( A0 s" s( ?
年度报告(包括长期稳定性数据的所有信息)。
3 O2 V- V/ T" @7 v7 L ! L; w# |+ @+ ^% A9 v
' h; R1 o7 t5 K! M8 P S1 v) w4 C" I9) r+ L5 C# z! P& _( G
- M, {) N9 v2 z) u' Y2. 2级变更, u$ m1 e' b c+ F W9 U
& ^+ y6 o' A" E4 ja. 2级变更的定义' p x l* c+ H- n( x: ~
4 {" ? [. d6 ]( N+ a/ Y( z- [变更为不同设计或不同操作原理的设备。混合设备类型的变更,如:高切应
* J9 {% h7 }0 k- u. i j3 D& L
) G4 v" l) |4 s) A力变更为低切应力,或者相反。2 M6 r; F. _* Q& A8 F0 G- G
* a: [4 O4 V0 {8 [/ x( h5 O* S6 J+ l
b. 试验文件ⅰ化学文件
2 N q5 l: @0 J# r& r
/ P) G+ \( I; b1 R9 p, l产品出厂要求。变更通告和提交更新的已完成批记录文件。2 W$ Z! m/ q! z2 R/ {8 U
% z# }* U; e$ o6 D, p8 w
已获得重要信息:在Changes being effected补充申请中报告1批3个月加速稳
7 L" r) N8 Z5 O% M0 y% D1 I . w3 C& \2 F2 j0 E/ h4 a; K2 x
定性数据,以及在年度报告中报告的首批的长期稳定性数据。
+ A5 r, [2 P2 R% C. I
! S& @# H2 \2 z$ Y9 f+ G未获得重要信息:在Changes being effected补充申请中报告3批3个月加速稳( _ r: z: g& d% o; G
" d0 j) u9 Y5 J, o4 \9 a e
定性数据,以及在年度报告中报告前3个生产批次的长期稳定性数据。
/ F: f' ?6 N$ }
1 ?9 N k. h d9 W+ X% Xⅱ体外释放文件9 x2 }4 h; e7 ^+ m6 X
0 `* z9 A9 i' D应进行采用新设备制备的制剂与同期采用原设备制备的制剂的体外释放速
& R5 \3 z% ]- I' r9 V# P& F
' e* O4 H0 ~$ U' a) O0 w率比较。应证明2种制剂的中位体外释放率(以斜率推算,见第VII节)在可接受
8 ~5 S/ Z4 Y& h, E* u 6 P7 n6 }) s( F) L2 x; G+ ^: T
的限度之内,试验过程详见第VII节(体外释放试验)。
5 s1 b0 q b! ?, @, S& i/ H! o k " e; L. w: G4 d
iii. 体内生物等效性文件无。
- Q( _% }' P& h- {* q# g + {( h# |( X- p, [
c. 存档文件
. j1 E) r2 w4 v
* L% G# C& k% q& p$ @' T) h eChanges being effected补充申请(包括加速稳定性数据的所有信息);年度报告(长期稳定性数据)。# M% }3 ^7 F) i4 J- P" r7 n6 H, i
$ H3 Z- Q5 ^3 e7 t$ T( s( y* S" i7 Y
3. 3级变更
7 d/ E: u0 a. p" u
( x9 [! g& ?6 g# M; }预计在本类别中没有3级变更。3 G+ u8 o0 J' g
/ Q4 P% y! Q0 D+ d1 I C: P" a& OB. 工艺
' I: F+ }. c( V9 ^ ; r& v; O/ q8 d# k2 a$ e
1. 1级变更' l& I5 U" X- f0 Z) j3 {3 L. b
/ o1 k- B# b! d
a. 1级变更的定义
+ M' h! N2 b9 }3 N) S
, Z J/ l& W7 y" p" L5 F; u工艺变更,包括在已批准的申请范围内的混合速率、混合时间、操作速度和( R! |5 @% S& X# f; W; n
" g& `9 m$ @ E1 S
持续时间的变更,还包括将组份(活性成份除外)加入到油相或水相顺序变更。
$ E+ l; g4 E" { # q7 S9 V- |1 N9 F
b. 试验文件ⅰ化学文件
/ E1 J+ k3 N/ P" [& \& g
1 X* r* \7 C+ S" J2 n+ a8 `产品出厂要求,其他不需要。
& z' I7 [' P H. |; v3 s1 a. v $ r$ a6 I6 v' E# v; q1 v
u, x: J! Z( z# M. Y109 n! F3 L) t0 m; l
% D% u7 ]% ]! b5 r
ⅱ体外释放文件
: v) h- H# L% ~: q
( y7 d# @4 q m. G9 s' ~: Z" J无。
2 c" F; I: d9 U5 z ! U5 V. b8 q; z$ e
iii. 体内生物等效性文件无。
! W1 b! }2 e1 m- Q. z 9 _* i, O5 H! H- p# x+ G
c.存档文件0 c3 b0 l6 [5 X0 j6 I9 {1 |
4 b* c6 s ?; d年度报告。$ N3 S3 Y" P6 o. g) B- Q
5 C: S3 \0 ?. H3 `! L
2. 2级变更4 r r% I$ Q( \( z. N3 k4 Y
2 y0 E1 B. c# d7 L' Y1 la. 2级变更的定义9 m7 ~( M, i+ O3 F# V9 G f
2 g. t* E; e' n1 q$ p; ]6 D: h! E/ m工艺变更,包括在已批准的申请范围之外的所有有关的混合速率、混合时间、
6 o) K5 r1 V3 s2 m8 m # b; y$ U$ x7 H5 W# s6 h, b
冷却速率、操作速度和持续时间的变更。还包括各相混合工艺的变更。: T5 P6 z, O' y
; R }( z6 w9 f4 R
b. 试验文件ⅰ化学文件) m, k+ C5 V( c/ J/ s8 T
4 t# z( b% i8 ?' q/ q
产品出厂要求。变更通告和提交更新的已生效的批记录。
0 T m) F) R2 L& Z( p ' n& T# D# h* h6 y. f# Q
已获得重要信息:在Changes being effected补充申请中报告1批3个月加速稳 }! c+ y& {" v: M6 ?& F
/ Z/ q/ o: w! e- R3 V, A9 g' V/ Z定性数据,以及在年度报告中报告的首个产品批次的长期稳定性数据。
5 a# F5 I( I; z" c H% X 6 p$ @( u6 M. l% g' S
未获得重要信息:在Changes being effected补充申请中报3批3个月加速稳定5 V/ v' R/ p: e& Y. i2 r1 E
+ o) q ]! W4 S6 `+ H性数据,以及在年度报告中报告前3个产品批次的长期稳定性数据。% Y: Q( G- U) @2 k' d! L' k
0 ~2 r% ?; C7 v: @, N% g. J' q
ⅱ体外释放文件
& r) ~) {5 q- _ : M( R6 g, j+ b9 q. j; i- ]
应比较采用新/变更工艺制备的制剂与同期采用变更前工艺制备的制剂的体
: f, a5 i6 l2 n2 G$ H* [/ S
" q! N% ~6 N1 X外释放速率。应证明用2种工艺制备的制剂的中位体外释放速率(以斜率推算,: x; I" \8 f0 t
' B( t: w! _# C9 E- b+ P见第VII节)在可接受的限度之内。试验过程详见第VII节(体外释放试验)。2 ~& X$ K, P: ^" h% |
6 K" c/ D1 m+ ~) s$ q' K- _iii.体内生物等效性文件
1 f- `% A/ U( s1 s- } 4 V# Z, \) d* i, j
无。
1 g1 S& Q5 H5 U6 \! D- L
" `5 x$ O& [+ i3 \7 [0 u* Rc.存档文件
' S; A6 s0 B% C8 {9 `( X' @ # J2 B0 b/ [' h4 V. J
Changes being effected补充申请(包括加速稳定性数据的所有信息);年度3 K9 N+ H: c9 ~( ^5 y3 }$ F
* f5 S: J% T) h. v9 ]' e
报告(长期稳定性数据)。. n7 K% J1 z* ?0 Z5 i
3 l: J! s/ W3 \: ]$ c
3. 3级变更& D& _4 v5 Z9 ]
) H0 n, M! e/ F, Z4 \# h( S8 M7 q
预计在本类别中没有3级变更。
+ t# z1 w+ Q* [7 i A) a# Q" [& [ 1 T0 ?; n7 z. V
V. 批量(扩大规模/缩小规模)
- I2 O0 j0 B6 ?+ X0 V: \! R. v 3 d' N d1 \: Q' B: D
本指导原则建议,用于新药申请(NDA)中关键性临床试验批次或用于简1 J3 A* M+ Q/ g
' X/ w( w5 x0 ~/ x _
7 } a) J6 l1 G6 k8 w7 v5 v- r4 I110 f' t' K7 Z! u9 w. }! m9 ]
7 H6 Y0 }- j6 Q( ~- `5 p+ y* H8 s1 Y
化新药申请(ANDAs)/简化抗生素药物申请(AADAs)生物学测试批次的最小 c4 j# f) i4 ~2 l. m( [
- G% M) S/ |, c+ L% `( e
批量应至少为100 kg或生产批量的10%,以二者中的较大值为准。如果与上述建议
# I; i$ ]6 M* q) r! L$ v4 i ( h% }* p$ H) E- \5 U/ o
存在差别,应与FDA的相关审查部门讨论。所有规模变更应进行适当地验证并可能
' s0 v6 h, Z) l6 I* S* R3 t
8 n. _ Y9 { \* y! ^. C由相应的审查人员进行检查。
: p2 a- z; w" D8 p1 [ Z 6 `* h5 p" f# E, P
A. 1级变更0 U# h2 C5 e% ]
* q3 W- i5 |5 J* |& t5 {2 o9 m4 V1、1级变更的定义4 `$ {7 w2 x) }
, ?+ d9 j* a {9 R6 U
批量变更,最多并包括关键性临床试验批次/生物学检测批量的10倍,如果
5 F: k. I+ U5 u3 |4 u
V; e; i' Q- I( l3 f! z- {& g4 d: ?试验批次和全规模生产批次采用:(1) 与试验批生产设备相同设计和操作原理;+ c$ m( T& [( A
: k# o- ?; b$ L8 [2 v(2) 完全按照现行生产质量管理规范(cGMPs)生产;以及(3) 相同的标准
5 x6 }, V- A: U9 q) V7 ]4 ]" x ' f. Y( w' ^' r7 E9 U
操作规程(SOPs)和质量控制,以及相同的处方和生产工艺。
% D/ D* `3 Z. P7 ~' d6 ^8 }
$ a' n- {4 I2 B& j9 A3 R* Z2、试验文件
. Z( g9 a! V- V% m+ T
z8 k6 w7 `0 B# M' a" ~6 x/ d; w) ra. 化学文件$ @9 z' i7 Q+ F8 u1 M/ A9 w
' p8 |0 X2 ~% b N. y产品出厂要求。变更通告,在年度报告中提交更新的已完成的批记录。稳定性试验:在年度报告中报告首个生产批次的长期稳定性数据。
. C1 T* V. E4 G2 _6 U 9 y% b* t X& d% w
b. 体外释放文件
/ J6 j3 z4 R3 A% z; b 1 ]. J8 p8 Y' D9 g
无。, ?( H, ?- k r5 l
2 ]) N! i" V( `3 qc. 体内生物利用度文件无。3 t4 E, |6 {: ?: Y
, O: |1 S- Y- L0 Y3、存档文件
; h* M: e" a4 D+ p) H! C) z - e. p4 @9 t* N! n
年度报告(包括了长期稳定性数据的所有信息)。# W. ~( ~2 B% a" k
9 ]5 T4 f3 ?$ |, \1 TB. 2级变更* ~7 |; \: S: y4 O. J9 d: Y. q4 Q
$ r: J9 R, C9 {% L% `2 M3 _1. 2级变更的定义% T# G1 x% o! X- m
4 a, k. X/ K. V批量变更超出关键性临床试验批次/生物学检测批次规模的10倍,如果试验2 q7 a- I0 r0 F
$ r5 |( k3 T& x; m4 y5 K% S批次和全规模生产批次采用:(1) 与试验批生产设备相同的设计和操作原理;/ j9 z3 O" K) y) m
- }0 T8 E0 w1 _# z# n(2) 完全按照现行生产质量管理规范(cGMPs)生产;以及(3) 相同的标准
8 X/ |4 C- S- P! \" Y( T f
+ q3 K7 y3 o9 P3 \1 q7 N. C操作规程(SOPs)和质量控制,以及相同的处方和生产工艺。) K: [5 f6 y$ {% m+ R1 x2 o
4 g! p7 ]8 v) r+ K2 B; u
2. 试验文件! Y2 F; {9 ^# Z
* h# G1 _7 q% s$ o2 ~% F: ta. 化学文件
2 L4 Y5 s& v* z
% A+ s4 [* Z+ Y0 ^( [: Q% H( V R产品出厂要求。变更通告和提交更新的已完成的批记录文件。
# z# V& r# } d1 r' ]+ }; ?2 ?
5 c2 _4 X% Y: I9 C8 f5 {3 u稳定性试验:在Changes being effected补充申请中报告1批3个月加速稳定性" I. ?/ m' }0 d' s/ ^9 Z4 V" u, k
! ]' s! p% _: W+ S# T" v" d$ c
12# X A5 N- A/ v8 q3 u
: s& B- {& a+ U! U5 Q/ R G8 ^数据,以及在年度报告中报告首批产品的长期稳定性数据。& l+ ~3 f* ]1 B, Z& c
* u) H% s. A! Q& g: a" M
b. 体外释放文件
/ F6 L; h, P- P; K) s/ M3 y5 g
( C3 H3 z$ ^3 F6 C0 N `/ u应比较扩大规模与同期生产的变更前规模的制剂体外释放速率。应证明2种
, K/ x! x& i3 F. I+ x3 w2 P. J , g5 G0 d# [" |
批量的中位体外释放速率(以斜率推算,见第VII节)在可接受的限度之内,试验过程详见第VII节(体外释放试验)。5 [$ Z$ A1 u+ A- `6 w; l2 O& ~! m- `$ S
$ j" E3 Y0 R" ^c. 体内生物等效性文件无。& K0 R& G( [. |( @1 m" P
! b! T5 K* ~5 ^$ d9 s3. 存档文件
0 H4 u) p1 w% p& \% g+ X- q1 f 6 m3 H( v& j0 l8 ~
Changes being effected补充申请(包括加速稳定性数据的所有信息);年度报告(长期稳定性数据)。! Q$ j5 |. A( W4 D2 X( [0 p, p: U( ^. P
+ M$ O0 _! E( _1 N* v7 J" P
C. 3级变更 T% W: i+ w! v, ^: K# f( I
) H$ b2 D+ T: r# k& \5 x4 y
预计在本类别中没有3级变更。 N D) V7 a4 |7 V2 F/ w+ `8 D( l
. [ C# H: c9 i9 `8 g: T" q
VI. 产地
; Z6 B; L" Q, v# ^1 s( G; g# G , p y7 V) a1 r0 f2 y) u
产地变更包括本厂或合同生产商的产地、包装/充填操作和/或测试地点的变7 D. P2 _7 ~ f4 V% O4 ~" H
" l% m. e) ]4 G# ~更,不包括任何其他2级或3级变更,如批量、生产工艺(包括工艺和/或设备)
- y1 K6 y; R) r6 u " I% W& X' h! D' c4 p
以及成份或组份。新产地应在过去2年内具有良好的现行生产质量管理规范
- f3 z* v- N5 b; P
3 Y1 t# ^/ ?2 c% ](cGMP)检查记录。
0 P/ g! ?" Z& S
- M' n1 A2 i/ ] L0 K% k对于单纯的分析实验室地点变更,如果新实验室具有的cGMP为FDA现行的# L9 ] Q6 J1 _4 K0 | r
9 }# ~' b0 y# t4 d1 j, G
针对存有疑问的试验操作类型的cGMP,可按Changes being effected补充申请申
- G9 Y+ [$ A$ F& P& ~ : g% x* ?" \% r. M
报。该补充文件应该包括:承诺采用与已批准申请中所用方法相同的试验方法;8 j# y- Z' r0 `
5 L0 u$ P& |# {) b5 a
检测实验室符合cGMPs要求的书面声明;以及1份测试实验室所进行测试的完整说
) [* P# d' i' f; K) q# T5 ?
( `# E: F; ^: }' ]/ `明。如果测试机构未接受所做实验类别的良好cGMP检查,则建议进行Prior approval8 n! y, v+ w3 @1 |, b! I
! M" E. \+ o. I9 Wsupplemen。单独分析分析实验室的变更不需要提供稳定性数据。- x- X9 _' e/ I# G1 `) O1 E, {
$ v4 |# z [' M$ o8 v7 \6 TA. 1级变更
1 o1 W# o/ Q8 f6 {
, z$ j! c3 U2 t; W( p5 \- M1. 1级变更的定义
" K8 m9 i5 G1 m. e, g, C5 I
& e% w) q: f7 q* `' Z8 }1级变更包括工厂内的地点改变,相同的设备、标准操作规程(SOPs)、环4 j2 ~/ q! K: e9 R: W7 j
7 o: A- o) ^1 H$ }7 a" g/ R境条件(如温度和湿度)和工艺控制,以及相同的基本工作人员,除管理信息和8 Y4 l8 M* Q' v' I" S% Q1 g1 K4 W
$ V: j+ w6 v: _
工厂位置外,生产批次记录未做任何变更。基本工作人员的定义为具有适当生产
: `' A& t6 H( @( s ) h. P- `8 N: a- L% _4 R' E
操作经验的已在工厂工作的雇员。
5 G- S5 e* N8 |1 y
2 C% x0 {' j8 R9 w3 V' I2. 试验文件+ B1 o4 o+ h; p+ U$ e
* H) U2 r" E& d8 y) B W8 f% s1 U' C2 W
13
; m/ R5 W7 I. O6 _/ q/ Y
* u' G& e& t C3 ~% z7 }a. 化学文件
. O9 R( f6 ~& L. ]2 R: q' l ( ^0 A( O! ?9 U$ w: P6 h
产品出厂要求,其他不需要。
/ Y" x- f% g. t4 E& w3 @
5 N' P0 T" G3 ob. 体外释放文件
F. N4 d# U1 i% |! P* C. s9 D " {( A1 t* K' ^% Y+ x" ?. V& ?$ x
无。
; i% V" X, X( u* @" e5 Z % f' B j# p* l8 t1 c0 y
c. 体内生物等效性文件无。
" f. a3 F+ Z: ^* R" X1 \
! n3 I$ h$ W" {! [+ F! v/ j3. 存档文件
* D. X, R. O1 g$ T1 U 1 U! h. n4 f6 o2 |' K5 B
年度报告。; I X0 E1 H$ U; p1 L6 \
3 B# `( p- v9 Q- n0 q% f
B. 2级变更
' {/ X% t4 l7 @$ A) k9 z$ E( P' ] - {& w2 L% \$ w [
1. 2级变更的定义- v. P3 C. l- p6 T" R% U
8 H. Q, D) J: c$ [( B9 H# j
2级变更包括在相邻厂区内,或相邻城市街区工厂间的场地变更,2个产地采
) k& l A* D m [0 U) n c" H) P& ~$ {, D( J
用相同的设备、标准操作规程(SOPs)、环境条件(如温度和湿度)和工艺控
: ?/ l. g% x' m , \ I- k8 V$ N+ ?
制,以及相同的基本工作人员,除管理信息和工厂位置外,生产批次记录未做任
( M% y4 p! w" X8 l) l! |3 b 9 U1 J* q0 l+ P/ H/ Q- i5 h: z A/ W
何变更。+ B3 G1 v. z. q5 _
0 K& r* g# M# L+ |" j& q0 J
2. 试验文件4 G+ {6 N/ J$ ~" p X
4 h% v' m/ S; fa.化学文件, _2 Y# o3 m# D
+ Q" q+ l$ e0 l* R+ Y. {0 K新地点的位置和更新的已完成的批记录。产品出厂要求。; T0 _& o* Q% x& R( ]8 ^
- C1 z# U+ b" N
稳定性试验:在年度报告中报告首批长期稳定性数据。
, H" {; [7 Y2 Z( J $ e' ?# C6 Y) R" D( Q. C3 X
b.体外释放文件1 }; x: v7 L( b! T+ b
* ] L; _3 a- A% G无。1 d* C( S4 P) |* J- f3 _' K
9 T, _$ H: p b+ ?' D4 |0 s0 V
c.体内生物等效性文件0 V+ N. e& p. P, ^$ o
5 H% P" X. Q- Q无。; P9 _; t* X$ q1 t! B& H$ ~
6 z" j: U- D ^7 c. k" e. {1 n; W
3. 存档文件
* S1 |( n5 F( [8 P; u. b 4 d. O5 r- a$ V+ `& X& n
Changes being effected补充申请;年度报告(长期稳定性数据)。9 P2 L5 e2 o$ A9 [8 N6 w: _ b
: D9 Z" O$ |( ~; gC. 3级变更; `; ^" J, c1 A, r: w+ q! w
3 o/ @1 Q% d o! D1. 3级变更的定义5 i1 X! P; z( i* n. P
8 W* |& O, B+ o/ F( U" v4 L3级变更包括转移至不同厂区的产地。不同厂区被定义为与最初地点不相邻
6 k! I U- p3 V! D, e % p1 B" f b* z9 c5 x& `4 p
的厂区或工厂不在临近的城市街区。定义为3级变更需要满足:在新产地的生产5 k$ I) {8 d4 n4 }3 a
* q V2 y. u2 t9 N. W6 ~2 k过程应使用相似的设备、SOPs、外部环境,以及工艺控制。不应对生产批记录做
0 h! \5 B4 {" v" p* r- q) B d
5 L' \: m: C1 B出变更,除非与其它1级变更一致。必要时,修改管理信息、地点和语言翻译。7 X; @/ [ t+ s* ` O: F
- c0 Z- c8 A, [! s8 q5 ^
& a$ l7 O! \7 E& O- X+ j14
9 A: z! L2 M9 W 3 B% q. m: q- x' x5 Y* ?0 G- U
任**合同生产商也为3级变更。( N; [6 t) |3 v# w# y
. M, Q; o: o# U9 N5 k" T& W2. 试验文件
4 m' D2 W- U2 T/ C7 K/ v2 l8 [
4 P' D9 J' |% h2 Ea. 化学文件
" H* @9 a' {8 Z, z& S : H9 K# Q$ [4 u
新产地的位置和更新的已完成的批记录。产品出厂要求。1 W6 l* x3 _) D; g9 B0 U4 A
1 ^# I$ F9 ~+ _" t# C; f已获得重要信息:在Changes being effected补充申请中报告1批3个月加速稳
w9 _! W9 b+ s) H. o3 F, w" X % ]0 x& w( v x
定性数据,以及在年度报告中报告的前3个产品批次的长期稳定性数据。
+ B" m2 _1 Q& @
P; O, _) _- L6 r( M0 }6 X未获得重要信息:在Changes being effected补充申请中报告3批3个月加速稳
0 n9 t4 ?% o# X, r5 a2 _" G
8 n; \) r' b" n- B* v定性数据,以及在年度报告中报告前3个产品批次的长期稳定性数据。/ t$ J% @: p1 Q" u7 w
! X; D% T" t- Z: h2 R- fb. 体外释放文件
3 W2 ^( k9 {. @/ b " b: O3 w3 W" Z8 m
应比较来自新产地制备的制剂与同时生产的来自原产地制备制剂的体外释 F1 c ^2 Z; l6 D+ V
7 {( o# m- v! s+ g# ^! D8 `3 z' U放速率。应证明在2个产地的制剂的中位体外释放速率(以斜率推算,见第VII节)在可接受的限度之内。试验过程详见第VII节(体外释放试验)。! t; ?1 M4 h& P3 U
1 u! V+ n7 P2 e" j! uc. 体内生物等效性文件无。) Z# R, z6 q2 x& t! R3 h v' F' M
' t/ Z: X; U' O; I- N4 C3. 存档文件1 _) S9 r3 k B7 S8 v# y' x
8 [& y- k8 \5 ^6 ?( d6 ?# R, ~& ?1 o% xChanges being effected补充申请(包括加速稳定性数据的所有信息);年度
& e. U0 }+ R" Z! p + T3 U1 ]7 `4 n. G
报告(长期稳定性数据)。
/ Q7 ?( E% X/ }! W
' t. ~* L1 Q" M0 rVII.体外释放试验
/ a8 t' h5 ?" N! j9 ?
: }" M P0 P& N. v2 a* l5 }8 }体外释放是几种可用于表示局部外用制剂(即半固体,如乳剂、凝胶和软膏)
v! T6 B; T/ p
4 o3 I0 F0 l( i6 `- n特性的标准方法之一。药品处方的特性或热力学特性的重要变更应表现出药物释- H& _. S7 N9 u! ^& ~4 N& m
file:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps17.png ' g+ r! k- h3 E
& R2 M. g h- o' S* x) j% ^/ c
放的差异。理论上,制剂处于释放控制过程中时,释放与时间的平方根( file:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps18.png t )! f) \7 L1 Q2 p& ^* l
# B+ {, P' w! Q呈比例,因为释放来自衰减范围内。
* [! x) T" |: p
# z! p, v4 W8 q# Y& B3 e局部用药制剂的体外释放方法是基于开放扩散池系统,如Franz扩散池系统,5 g5 n5 k& v0 [1 \7 q9 c. g( J
$ I; X2 e" s m# c# d/ {5 y+ V
通常安装了人工膜。受试品置于扩散池的开放供体腔人工膜的上侧,采样液置于
5 S8 }/ N0 x7 \+ R# s) d! A. }
6 ~3 F5 k5 X+ G0 P& f' E接受腔人工膜的另一侧。通过测定连续收集的接受液样本,检测药物从局部用药
3 | e7 `/ K2 J8 V: |. X/ O ( t8 d& H: ~6 A; a( D* {0 N' n
透过人工膜的扩散。应进行适当的体外释放方法学验证。收集样本可以是自动的。8 @: k) Q7 k& V# ^* T
% R" ~( r K+ B+ n0 l从接受相转移出供试品可通过高效液相色谱法(HPLC)或其他分析方法测$ X$ Q. h) D, v1 t
+ s8 x- I! J; t9 S
定药物含量。用单位面积释放的药物量(mcg/cm2)相对时间的平方根绘图生成
4 z! p+ d/ ]# h: M9 D2 T& t
0 ~+ n. S% {: i- m( h E7 g一条直线,其斜率代表释放率。释放率的测定具有剂型特异性,可被用于监测产
1 `$ |$ w: P$ C' l$ W! B 5 t$ v- ?0 ]& C. N4 m' K; ^
品质量。按照本指导原则的规定,应进行生物学测试批次或现用生产批次的释放0 t7 G- r- l, _- l
! X2 i1 V4 L8 h b! q7 i2 G/ l; y4 B7 d6 Y2 Q4 w
15* h; `- f% \# H% \
; Y; a5 j* q4 g' O9 L
率,与在本指导原则中确定的变更后制备的制剂释放率的比较。2 ^/ i0 F! y4 p/ ?$ e( m. \ a
# Q- p2 s( J6 D: u' N
以下总结了体外释放研究设计的一种可能。鼓励申办者对此处列出的参考文. T9 Q. C7 `2 d$ G6 R& _
, }$ G3 K+ L/ {( r- l/ r献进行评论。
/ A) Y9 ]. ^8 J c+ k- f9 _# U: @ 8 `1 r/ |4 d5 U
扩散池系统:7 _: E2 h5 ?0 p
" A5 v4 t$ ~ _' @, g7 x' s8 b- vz 带有标准开帽式毛玻璃样表面的扩散池系统,注入孔直径为15 mm,整个系统直径为25 mm。
; V/ |. } C/ ^& _, _, F- x3 t" N5 u 6 L/ l3 l: f3 v3 V6 ]
人工膜:1 f r# G' C5 Y6 M' D/ e
8 H2 x5 g% Y$ t1 |' Lz 适当的惰性和商业化的人工膜,如:聚砜,醋酸纤维素/尼龙混合酯,或聚四氟乙烯70μm膜。大小适合扩散池直径(即上文提到的25 mm)。
4 Z+ _$ A7 j5 }4 i
5 l( K0 m6 ] }- h7 l+ M% W接受液介质:# ~2 v; S6 |- x
) [" H9 k/ H3 c1 m
z 适宜的接受液介质,如水性缓冲液用于水溶性药物使用,或者含水醇介质用于水溶性较差的药物,或其它经过适当调整的介质。' T F, {4 ~' k( w/ v: W6 K8 {
f1 j4 \$ A( c
样本量:2 b9 O6 F" H' P, N7 x
2 W7 C- [5 O9 H |多次重复(建议6个样本)测定皮肤局部用药的释放率(特征)。样本使用:; h9 O' a; k( H- d
0 d5 l4 e/ @' b7 f* [2 C, o' Q- A
z 将大约300 mg半固体制剂均匀置于人工合成膜上,保持封闭状态防止溶液蒸发和成份改变。这对应于没有剂量限定的情况。
% ^5 q% ]" R5 c K" \5 T5 L ' P3 a2 v1 C8 \* k0 u' Z7 Z; W
采样时间
: S6 ~7 }$ o/ H7 Q% m7 C: k
' c' A* p8 j5 Q; M! mz 建议在一个适当的时期内多次采样(至少5次),以便产生足量释放的特征和确定药物的释放率(在为期6个小时的研究期间内,采样次数不少于5
3 W2 i' M2 R, h% M: B3 j* q 4 r& Z+ r9 n( ?) L, B
次,即采样时间为30 分钟、1、2、4和6小时)。不同制剂的采样时间可能不同。
% ?2 i2 [7 R! G* w7 M8 `5 g
( M. V1 P' @9 M3 O& {% G在每个采样点取样后补充新鲜的接受相液体,以便在整个实验过程中人工膜的下& e0 M3 P% W e
3 r! q' U" g7 w- }表面始终保持与接受相的接触。
3 D" q$ k. V: ]. l: O
( W; i3 `- y1 Z7 G样本分析:
7 X d8 [. {6 ]. L" M
5 p# S& e+ F1 V: {0 p# r1 |% Uz 应该采用经过适当验证的特异的且灵敏的分析方法分析样本,以确定药物浓度和药物释放量。9 n# F8 N* p' A6 f
' [2 T( R, B3 a4 Z; S0 Q ~% j+ a9 `
体外释放率:
. i5 I3 z9 l1 l2 `' G, e+ k * M4 H0 x) }3 ~% B- ]; j7 O5 a; D, w
z 用单位面积释放的药物量(mcg/cm2)相对时间平方根绘图应生成一条直线。直线的斜率(回归)代表制剂的释放率。对应于1小时部分的X轴的截
) A1 B3 n9 M9 F' q% H
; S4 X( V, z) z2 D: I1 L/ e, J; `, j距是此类曲线在正常状态下的特性。
( H# C6 c+ _3 T
# \7 y9 f# [/ j" @! Q+ {0 H& J' s- r1 H5 F
16
$ A) [% u1 ^/ P2 X6 j
4 W- Y3 P( p% u! c" E/ M8 t释放率(特征)比较研究的设计
; d! j! F* v. ~' s+ p; L# V; v ( t7 k# s3 u m0 X' w; y
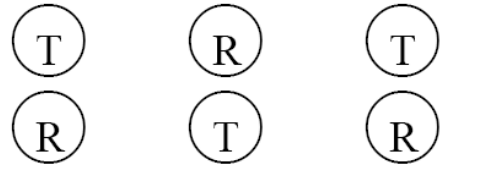
z 典型的体外释放测试设备有6个扩散池。对于测试设备的每次运行,应该将比较研究所用的2种制剂按以下分配:
3 F) S+ P# H' y m0 G( U1 P ' Y7 G9 L$ W2 T
或者4 A. b9 U( d, D" u. F- `" e1 w
7 {$ A' G2 T$ G& u
* Z% X. B9 z$ ]" v1 B, m! k2 D: Z3 t& t
此处,T代表变更后批次(受试制剂),R代表变更前批次(参比制剂)。采用这种每次运行中包括2种制剂的体外释放的方法,有助于消除运行之间存在系统差异,确保无偏差的比较。
7 d8 q. S6 z7 y/ @/ @" p
- V+ f! W+ S) ~( O/ e5 h: uz 将制剂分配至扩散池的选择(即,将变更前批次还是变更后批次分配至设备的“左上角扩散池”)可以是系统性的(即,更改每个连续运行的模式)或随机的(即,投掷硬币或使用某种其他的随机机制)。3 c- @ \4 A# p4 U- g: v
2 K- |$ y. Y3 } k% P1 ]
z 如果使用非6个扩散池的非标准化设备,仍应遵循在同一次运行中包括变更前批次和变更后批次的原则。如果非标准化设备仅有1个扩散池,应混合运行变更前和变更后批次,而不是在获取了一种制剂的所有检测数据后,再获取另一种制剂的全部检测数据。8 U' S2 H- R y
3 D/ b8 M' l1 \- x' p1 w6 ?$ O体外释放比较试验的详情
( a* k5 E8 |- j0 [% c+ o
0 X+ G) S* t6 k3 J# V) m2 oz 体外释放比较应该分两步进行。7 n: _& A5 H, _
5 u1 u% ]& E6 [. K J' K
第一步,体外释放设备(6个扩散池)应该运行2次,变更前批次(R)和变
. V9 J5 m+ P( N. @ R
3 M) ^/ \. s" k# H更后批次(T)各产生6个斜率(估计的体外释放率)。应该计算变更后批次的中
) o2 a( `( m+ H0 i " |7 w1 ?9 h2 a) g2 |
位体外释放率(总体中的)与变更前批次的中位体外释放率(总体中的)比值的
2 m) ~6 k* u2 [/ C1 Z 1 ~9 b( G6 ]& i9 J$ A6 o
90%置信区间(见以下描述),用百分比表示。在第一阶段,如果90%置信区间
7 C8 e: @( Z4 [! x
& O2 r7 |( P3 C8 r% m m' X# A& A7 w3 F+ h8 ?0 e) [3 z
17) |9 }% B+ N/ N0 N8 V
& I& y/ S6 L* w0 e
在75% ~ 133.33%的限度范围内,则不需要做进一步的体外测试。
1 g4 J, K5 v' y# M7 q. u
/ u: q; X0 p; j2 w1 P如果第一阶段的测试不合格,则应该增加4次体外设备(6个扩散池)的运行,! Z+ j, E" y+ a8 l
" O P$ l) i1 f- T3 x变更前和变更后批次各增加12个斜率,或共18个斜率(包括第一阶段的结果)。
+ k4 B) I" b+ o: n- x
4 E3 l3 _) I; e. `每个产品共采用18个斜率(包括第一阶段的测试结果)计算制剂的总共的90%置
, L9 ~2 H( S2 K) G& e. F# W! [
7 h+ a/ D! l' F( K5 _信区间(见下文)。在第二阶段,上述90%置信区间应该在75% ~ 133 .33%的限9 l; I8 t) J, D# P/ K6 f' N
& a2 O* ]; G: k2 H K
度之内。
& G: e5 [% t- V* L1 B 1 i: v2 f1 c3 K/ U6 }
置信区间的计算——1个实例:
( ]7 n& W* E9 ` : ?; D6 a: p m* G
z 由于预计此次测试时会出现极端值(例如,由于制剂样本和人工膜之间存在气泡),所以建议使用易于抵抗极端值出现的非参数方法。在以下实例中描述了计算方法。
" @: V/ U' t: v; z+ i& `( d
+ l! _' ^+ i A假设第一阶段获取的斜率数据如下:2 W5 V3 Z, j& z" A6 a. c
; p2 Q% S& ?1 J- _
' ~1 Y! J/ I6 f
/ R5 Y8 G6 j! ^6 p1 D! x+ Y变更后批次 (T) 变更前批次 (R)
6 @5 A! a; J V2 N1 Y( M ' e$ f% ]4 ~: U6 B& X' V' n) _
置信区间计算的第一步是产生36个(= 6 x 6)单独的T/R比值。在下列表格
( r4 ], n& |' t2 x* M$ H1 M7 x
$ Z* C# G6 w2 D中给出了各T/R比值,变更前批次的斜率(R)列在表格上部,变更后批次的斜率; U# x" w' L8 Z5 v! J. r
: U+ r6 V+ a+ R: h3 e(T)列在表格左边缘,表格的主体部分给出了各个T/R比值的数据。
% r3 M8 R \3 t) ^2 S
8 u, [$ j! s# ?4 ?% c! B2 n
" g$ b" B9 O+ d" O0 F| | 1.1331 | 1.1842 | 1.0824 | 1.3049 | 1.0410 | 1.2419 |
| | | | | | | |
| 1.3390 | 1.1817 | 1.1307 | 1.2371 | 1.0261 | 1.2863 | 1.0782 |
| 1.3496 | 1.1911 | 1.1397 | 1.2469 | 1.0343 | 1.2964 | 1.0867 |
| 1.4946 | 1.3190 | 1.2621 | 1.3808 | 1.1454 | 1.4357 | 1.2035 |
| 1.4668 | 1.2945 | 1.2386 | 1.3551 | 1.1241 | 1.4090 | 1.1811 |
| 1. 1911 | 1.0512 | 1.0058 | 1.1004 | 0.9128 | 1.1442 | 0.9591 |
| 1.2210 | 1.0776 | 1.0311 | 1.1280 | 0.9357 | 1.1729 | 0.9832 |
| | | | | | | |
/ r& J9 ^: ]8 @+ u/ H置信区间计算的第二步是将上述36个T/R比值从低至高进行排序。
. Q# O3 \( f) g" s; [/ g7 d, }
" I+ V9 e$ m7 j5 h" _; I) i* k2 @, N$ M8 E. J
18: X r. S$ E* _) q I
1 V/ n* Q n7 A
0.9128、0.9357、0.9591、0.9832、1.0058、1.0261、1.0311、1.0343 . . . 1.2863、
/ W0 I. d$ E, l" O' H
+ X/ J& a+ p% Y! ]; [; P: h K% ~1.2945、1.2964、1.3190、1.3551、1.3808、1.4090、1.4357。& a% |2 T- G* A
! ?9 c$ f+ q% p5 o+ F- L: r
第三步,上文中排序的第8个和第29个比值分别是T的中位体外释放率(斜率)
4 ^2 W! o5 d C9 ]! q, z- x
- E0 r0 f. e" F% Q: Q与R的中位体外释放率比值的90%置信区间的下限和上限。本实例中,90%置信
, J' ?& m2 @- D. n4 x5 h9 Z
$ {: ~3 i. ^" V区间为1.0343 ~ 1.2863,或用百分数表示为103 .43% ~ 128.63%。
4 ?4 v, e7 v/ L% e5 V& ]* w
6 W7 Y w6 x" t9 Z6 l5 D& z* q由于90%置信区间在75% ~ 133.33%范围内,所以本制剂通过了第一阶段测试。, u) W( i i9 J8 ^
& o, u' M w4 c: q如果第一阶段的测试不合格,则应该增加4次体外设备(6个扩散池)的运行,
. T7 F5 [0 M2 O
' h' i2 `+ X9 X+ c4 n9 |3 U变更前和变更后批次各增加12个斜率,或者共18个斜率(包括第一阶段的结果)。
7 |- J. v9 _ P" P8 Q
4 d% c ]" B" o7 a8 g应获得全部324个T/R比值(= 18 x 18),并从低至高进行排序。显然,即使不! Q0 ?5 ?; F5 \3 t4 q: w
2 F. p" o; J+ P5 l: S
厌其烦地手动计算了第一阶段的数值,但第二阶段的数值无法进行手动计算。应使5 D2 z2 r0 W; j" n6 m8 K7 d
& q5 x' z1 ]7 Z, M' F5 E; }) g
用计算机。
2 U0 t9 Z; D! y' m3 s9 O 4 T/ |2 S' j7 h2 ^# S9 I
第二阶段,位于第110位和第215位的比值分别是T的中位体外释放率(斜率)
& h4 G9 ]% V' P2 }/ T$ I . @! r+ r S5 j
与R的中位体外释放比率比值的90%置信区间的下限和上限。如果90%置信区间2 U3 u$ b$ E8 `3 r8 m
$ o0 ~9 r5 a( u4 E- B在75% ~ 133.33%范围内,则本制剂通过了第二阶段测试。2 x5 x. ?) q2 Q: A9 u9 {: L* x
8 D( Q" \4 L! L/ i! H9 O X }体外释放比较测试的进一步评论 x) U" j0 ^; b. F( C
; s9 d6 p" W3 H' F% W" L
z 上文描述的统计学检验是基于与Wilcoxon秩和检验/ Mann-Whitney秩& D: G4 Y& E2 i; A
7 H; p( p( B8 J. T4 E$ X- C: \和检验有关的标准置信区间计算方法,适用于计算对数斜率。置信区间计算方法的参考文献包括:3 ?! ~8 G1 p: n& [
; `2 A% v L3 ^3 ^! Z, _/ k
Conover, W.J., Practical Nonparametric Statistics (Second Edition), John Wiley & S**, page 223ff, 1980.) d8 U/ H' J" o8 T1 s( I6 W7 c
: {+ B! R" F) |* y4 PHollander, M. and D.A.Wolfe, Nonparametric Statistical Methods, John Wiley & S**, page 78ff, 1973." o, J5 o" V- S4 E
然而,如上述实例中所见,没有必要为了进行上述检验而精确计算对数值。
4 j- F/ t b, n) B0 d& R ! C; j: U) [ ~+ h$ u
z 上述实例说明了全部数据的情况,即第一阶段中每个批次有6个斜率,并且如果必须进入第二阶段,则第二阶段时每个批次有18个斜率。如果斜率缺失,则需要调整计算方法。例如,如果第一阶段时某一批次缺失1个斜率(如果是变更前批次或变更后批次的斜率,则无关紧要),则将只有30个(= 5 x 6)T/R比值,而且90%置信区间的范围不再是第8位和第29位的T/R比值,而是第6位和第. u/ u3 i+ f2 E# ^& D, e+ v
8 }( `0 ~" J) s( X25位的T/R比值。如果任一测试阶段的数据有所缺失,则应采用参照统计学教材或$ N& g7 R6 A' s
3 b& O' e$ t/ m5 T" g- M1 {
h4 C2 h ^3 i
19% b) s. ^0 | a" _/ f+ f
( m/ D+ A+ E8 Z
咨询顾问、或者咨询CDER人员校正计算方法。
B b* i; N7 K) M* h- j
1 I4 o' [6 G! h: } e: _" Sz 上文描述的统计方法没有考虑测试的程序块结构(即1次获取6个斜率运行数据,而不是一次获得全部数据的事实)。通过以下进行判定:
0 i$ R" [6 p( e6 p& u) c/ ^" @
: D# c0 W, ^# j1.在提供药品审评中心的体外释放数据中没有证据表明重要的批次间效应。
+ b/ b7 p0 G5 M; P7 @* x8 N0 Y
7 v( r0 M0 V: |6 a: K2. 所建议的每次运行包括2个产品的实验设计将有助于在出现批次间效应的情况下确保没有偏向性。
+ H! a! y& A! O1 T2 s* o) n
0 A2 |' @4 A( _; I. |8 @6 F1 rVIII. 体内生物等效性研究
! Y# _1 `; c; m3 M
" d, S% X# X/ L5 V6 N4 C, {5 f半固体剂型体内生物等效性研究的设计取决于药物和剂型的药理学活性。以
0 ~5 d5 G% H. }6 D; `4 U' F" _ . p/ [9 e0 {% ^& R
下为此类实验的一般性简要讨论。8 ]# }. J& X7 b. l' t
0 F" Q6 u7 H9 d D" i- U7 S4 t2 C
目的- w g( c- ]- G4 w( N D
0 A1 Z( v( e; v; H$ o& a$ S W
进行与变更前产品或与参比制剂(RLD)的比较,以证明本指导原则限定的
" S4 j/ b' m+ j % ]) h) r' S9 W$ X" k* P3 X+ f+ Q
变更后生产的药品的生物等效性。
6 u: _ G; h" A + ~# \' q9 h/ _1 P& f/ f, S
设计, g: v7 S; K9 S( T6 Q! G) ?3 H( B- y; T
0 t3 C& U) _3 g5 q
研究设计依据活性药物的性质。生物等效性研究可以是类似在糖皮质激素类3 h5 L2 D3 @6 G8 `0 k! X
8 h! m1 n# r% d% F中所用的一种比较性皮肤褪色研究(FDA, Topical Dermatological Corticosteroids.
s( I2 B# \6 O( h7 i - h8 \" n) `! T7 d
In Vivo Bioequivalence, June 2, 1995.)或者是用于皮肤外用制剂的一种比较性临
; d/ O c- W1 Y" z i9 ]
6 c& X2 b+ A( I6 r. V床试验或其他适宜的经过确认的生物等效性研究(如,皮肤药理学研究)。4 D$ A2 a" r$ q7 C9 b1 I; [5 v
) h. I; C& h/ E, F7 {分析方法
3 _ T; S; Q7 B' g. t0 l 6 r/ ~$ h. V6 [' ]0 X
所选择的测定方法应能确保特异性、准确度、日间和日内精确度、标准曲线+ _* O* c" a0 D/ `0 B
8 U6 `$ `# P, u( F9 H的线性,足够的灵敏度和回收率,以及样本在与分析方法有关的保存和处理条件
' A# V+ d' C% k; \8 Z* }% f6 S
8 H' ]+ j5 m% F4 o' `7 u下的稳定性。9 u6 Z% C- {( T) N
6 _5 @, F( E8 o
5 |- E! E" T$ `% N8 @: F6 S) m
6 Q4 b3 a! c: B" l# U' c8 M203 m. b' l' B' g6 X
" ?% ]$ k* b9 `5 K+ X; V# D6 k术语解释2
) @+ P' X# c+ e0 f0 K- i1 X; _- n
9 Y" y! T7 O$ a! y" k, D5 _已批准的目标组份:在已批准的关键性临床研究或生物等效性研究中所用药% O4 B0 P( ?% c% j
' l# v; j- m3 u1 ]- B
品的每种成份的组份和量。 o8 ]/ V. d* T9 f1 I
" n& v# `: W7 M w" N
批次:根据在同一生产循环中各生产顺序生产的并且预期在特定范围内(21
6 Y& i7 d2 ~% k, A$ r
; c3 n3 D+ W& s2 S7 a9 NCFR 210.3(b)(2))具有同样特性和质量的药品或其他材料的特定数量的产品。
0 G! q2 q/ H0 U- ^. a # A, X" P0 c7 _& r) j# j
相邻厂区:相邻的或相连接的场所,或者在临近城市街区中的一组建筑物。6 X2 l! t9 [# p9 D$ d8 s2 P
3 {! ~! M2 y. C
乳剂/洗剂:含有完全溶解或混悬原料药的外用半固体乳液。洗剂一般黏度
& U0 M& v% z* T" l+ [+ C6 F! n0 W3 a
- B! k- Q! V4 A9 c' h. Z+ y4 D1 J1 q较低。
% }, T% u: e$ e5 K% `+ N 5 F9 N u# |( P
稀释剂:制药处方中通常被用于定容或定量的溶媒(如,水,石蜡基)。
/ }6 v. R" m2 `' T. T3 C 6 ?7 v7 m# O$ r' A
药品:药品指的是上市包装中的成品剂型(如,乳剂、凝胶或软膏)。制剂
( `2 L( H; u: o$ [
! d. @! K) B; u6 L3 P/ Z也可以是含有一种原料药的成品剂型(如,片剂、胶囊或溶液),通常但并非必
$ {! {7 [+ P* s5 | e0 Q, @ + L& ~0 \/ V7 \; H- l
须含有一种或多种其他成份(21 CFR 314.3(b))。! F8 {& j+ [ n- C, D
u5 S3 s% a6 k+ ^
药物释放:药物从其制剂中解离经透皮或全身吸收而使得药物发挥药效。
8 Z& o$ M. l8 y6 y/ T/ i# }+ B( b
: E9 U8 R8 V4 Z3 h& w8 c- P& m原料药:预期能提供诊断、治愈、减轻、治疗或预防疾病的药理学活性或其- N1 {; D& I2 r6 _9 C
3 E& t/ ~* r3 B4 C. k他直接作用、或者影响人体结构或功能的活性成份,但不包括用于合成此类活性/ N, ?& v: E8 k4 A' f+ _
" P. f2 T% V: O; r" c3 o# R成份的中间体(21 CFR 314.3(b))。" _" o, @9 R5 C- o: n: o g
. r% p U) X) y; a1 d
乳剂:乳剂为两相系统,其中不混溶的液体(分散相)以小液滴的形式分散$ M6 R+ Z: C" j, F1 R6 a' k, a
5 t* H0 h0 c7 U# A# @! x- e5 u在另一种液体中(连续相或外相)。如果油相为分散相,水溶液为连续相,则该
; w+ m; Q4 l: D& e) b6 q
6 i4 M7 {4 L& L# j; K系统被命名为水包油乳液。相反,如果水或水溶液为分散相,油或油脂性材料为0 W- s8 z: ]7 |6 P9 R
$ d+ H8 W' c; w/ l% p* T' D
连续相,则该系统被命名为油包水乳液。乳剂通过乳化剂保持稳定,可预防小液
' @- a( x% j4 [" P/ W' \
6 a. b5 }; V* J. z( e+ B滴**和合并为较大液滴,并最终进入一个单一的分离相。乳化剂(表面活性剂)' W$ S- ~8 Q! S: Q/ T& Q
2 p- o+ a( Y o+ l, M; V' M- B9 `
保持乳剂稳定的机制是在液滴和外相之间的界面浓集,并在粒子周围提供避免融, ]5 R/ q" Q5 P H+ l& v- K
3 y7 Z) _ {) x7 i) T( m V
合的物理屏障。表面活性剂还可以降低各相间的界面张力,从而在混合时更易于% ~8 h& ~' y" u2 f: q- ]
- v$ x D, ]% w- f( R3 c ^
乳化。乳化剂的本质是预防乳液微滴融合或延缓乳液微滴融合所需的时间。乳化
6 K9 F) ~% b3 h; g6 n
: W1 a9 N0 X* A7 L$ ^3 _2 r/ J7 O) J是形成乳液的过程。乳化可以包括一种液体融入另一种液体从而形成乳液或一种
9 a: K) ?1 z2 }3 O9 A
! r5 `. e, L0 E' m) d& ]3 X# s气体融入一种液体从而形成泡沫的混合。& x% w a+ A9 g: Z* t$ V
$ E% ]# b8 x7 |# C0 E' Y
处方:制剂的组成成份和量。6 X1 ^, _- i$ k3 T3 j
file:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps23.png
0 C# Y7 \( g5 r# w" v
, Q9 n" V$ }& N: D1 Y, m7 a 0 i2 p: G' ?9 b) h- p
2 见研讨会报告:液体和半固体分散系统的规模扩大。G. A. Van Buskirk, V. P. Shah, D. Adair, et al. Pharmaceutical Research, 11, 1216-1220, 1994,
2 ^. |' V) _( B6 @9 _+ o5 s1 g ; Q+ p# P3 e$ ?9 V) H) J
5 }3 E4 o7 }/ N0 C5 N9 p! n0 W, C21& N; Y$ i, U$ ` _9 b! c
: w: \. D( V) B i凝胶:一种半固体系统,其中,液相被限制在一个三维、交联基质内。原料( c0 q5 E0 w! T
+ d& }' @. h9 X8 N! [, _ n8 A2 w& i; |
药可溶解于或悬浮于液相内。; C, Q' d& h) y% }7 y4 K
2 N& E* t- A9 ^! ~0 O
均质化:一种雾化方法,一种液体借此进入另一种在高压(如,可高达5000
1 B+ I! v. g- X( x0 s " _0 D( c! O) w
psi)下被压制在精细阀芯和阀座之间的液体中。
/ `$ A" {+ O5 E* ]! p
5 Z' k% G: @: U% i内相:乳液的内相或分散相,由在乳剂中存在的微滴组成。5 N( f1 [- W# n/ S& c2 T6 R
$ q2 D$ Y% a9 p体外释放率:活性药物从其处方中释放的速率,通常用数量/单位面积/时间0.5表示。
. r+ m+ R% g, X8 @7 W 8 ]4 y" R" M5 x P. l, @
软膏:一种外用的油质半固体制剂。典型的软膏基质是凡士林。软膏中所含
0 e: T8 J# J! g6 P( G" C + {0 ]: O/ l& A# r2 B
的水分不足以使之在室温下分离为两相。水溶性软膏可用聚乙烯乙二醇配制而; P* A3 R( k4 G7 B( y6 J" ?# b
; ^5 B" E; W* s成。4 c/ o2 r' ~5 Z+ X2 p
1 Y+ J n' j/ B0 v A2 f- h
中试规模批次:制剂的生产工艺为完全能代表和模仿计划用于大批量生产的工艺。* {. h6 j; J: \3 S
- r, b: s$ a5 p3 C, g8 A+ \' ]2 x防腐剂:一种被加入制剂中用于预防或抑制微生物生长的化学剂。) y1 W% m$ j I1 l
6 Z/ l. b) t& `3 L8 H# t. @
工艺:用于生产制剂的一系列操作、行为和控制。
6 g5 G4 c6 }# c 7 ~! G( N8 N* D
扩大规模:增加批量的过程。
" {* J! L( Q" B/ s ^* m$ [4 n ! m7 o' V, L* R5 L
缩小规模:降低批量的过程。
1 G4 q1 h9 ]% l x: D' C
5 w& c! {% W9 Y切应力:引起或倾向于引起物体的相邻部位以接触面平行方向相对滑向另一. E% {' a" c; s# [8 k' q: D& l
( n2 C( X3 y) D1 P. C! h1 G) {( r
个部位的应力所形成的一种张力。在乳化和混悬过程中,通过匀浆器或其他研磨 v: p3 R. d0 H
5 o9 _5 c! i: g( O; Y; q设备,可系统过渡时产生切应力。, J- I* ?6 \2 {% r4 f
7 M: W4 l$ D; \5 Dz 低切应力:通过混合和/或乳化切应力产生低张力的处理作用。 g. K" ~, O( Y" g" v, c+ i' {
, `) c) B& `! P# c |$ v
z 高切应力:强烈的处理作用,在混合或乳化时在制剂上施加强大的张力。匀浆,究其本质,是一个高切应力的处理过程,可产生小的和大小相对均匀的乳化微滴。根据操作,研磨和搅拌器可被归类为高切应力或低切应力设备。6 \7 {! p0 M) D) m* Z/ _9 @
) v. v; S3 f8 i重要信息:与制剂稳定性有关的重要信息,可能出现在新分子实体上市5年+ n& \4 E( a# \" s/ D7 n
8 T4 S* G( z5 J' ^: G% P$ }) i
后,或者新剂型上市3年后。
# M9 a! L" R* \" Z4 Y; h% a 0 j3 ?& I' C, j
赋形剂:一种参与形成结构基质的辅料,赋予软膏、霜或凝胶等剂型以半固
' H+ A$ h8 m1 I- r! q0 u
6 b( R! g( o) Z5 `3 k/ \体特性。实例是凝胶赋形多聚物、凡士林、某种胶状无机固体(如膨润土)、蜡6 ^+ i% D7 {, ?" ?. m# K T
L* L9 W, V5 O
状固体(如,鲸蜡醇、硬脂酸)和乳剂中所用的乳化剂。
, t/ a! T, S* }( l+ ^ 0 Y0 a5 N6 i }
规格:规格为原料药的浓度(例如,重量/重量、重量/体积、或单位剂量/
$ |+ h+ k# T! ?
% d6 V+ m. Q% _; m6 ^7 z( L+ o$ ~6 K体积),以及/或者效能,即,通过适宜实验室检查或通过充分开发和对照的临5 y, s- ~/ F0 y; Z6 C* O ]5 U( j
: S# H- }( d1 V4 P& _" P) u h8 J2 R" n/ K. m5 c; j; s* L
22
: ]. N# F# m' @/ |9 q2 J
5 G8 `6 n ~' H }, v9 o- s床数据所表明的药品的治疗活性(例如,用参照一个标准品的单位表示)(21 CFR; z3 `8 Q3 z# P3 S2 I$ z! ]
/ p' N& _# ^( Q" {210.3(b)(16))。对于半固体剂型,规格通常用重量/重量(w/w)或重量/体积
0 V0 f/ ~' P3 W. R3 s
. z$ v. H6 O# z9 T7 a; N(w/v)百分比表示。/ {+ G W6 x: }6 j/ g
1 ^' p) q% Z3 ^) ~1 E! S% |
混悬剂:一种加入混悬液用于控制活性成份沉降速率的辅料。; t& ], F* ^& l L) o
) P" O* k, B3 T技术等级:辅料的技术等级因其技术要求和使用目的的不同而不同。技术等7 u y0 R: }: N
" ^0 U2 Z7 o- S+ s- [$ D# s. w
级的不同在于:(1) 技术要求和/或功能;(2) 杂质;以及(3) 杂质谱。. R1 H* ~3 e4 m; n% n
. P* g* n8 j, F% E7 N
验证:一种确定证明依据的过程,该程序可高度确保特定的方法或测试可始/ v: o& o) C! y7 g
$ E& W/ ]; _) [. y
终如一地生产出符合其预先设定的技术要求和质量属性的制剂或测试结果。一种! g$ X& l1 n+ |% X: [
8 a; a& R# D9 L; V5 V4 C- U
有效的生产程序或测试是指已经被证实了符合声明或描述目的的生产程序或测
, h4 W D' t. C# r" u * l! Q* J- L+ J2 }& ]3 d; z
试。程序验证的证据来自数据的收集和评估,最好是起始于程序开发阶段并贯穿
! N' B4 {8 k2 B5 d" z
( }" W+ L, U# x1 ^" D5 ~1 b于整个生产阶段。程序验证必须包括程序合格(材料、设备、系统、建筑物、人3 l5 P! a6 F# a! X* S* T
5 ^! |3 [, ~$ D- u$ P. m6 Z
员合格),同时还应包括控制重复批次或运行的全过程。7 l6 t+ ]! j, _5 K) K
* @/ d) t0 P# _0 L8 p+ q2 J3 M ) `+ D, ?3 W/ v' ?
参考文献4 K S3 @! s6 L& R' H) B S( V
9 Z4 I% G; j4 |! ]1. Shah, V. P., J. Elkins, J. Hanus, C. Noorizadeh, and J. P. Skelly,"In Vitro Release of Hydrocortisone from Topical Preparati** and Automated Procedure," Pharmaceutical Research, 8:55-59, 1991.1 P f# }! r, ~, Z' w( q9 k
, X3 u7 f+ E' Q% r" \) A! X
2. Shah, V. P., J. S. Elkins, and R. L. Williams, "In Vitro Drug Release Measurement of Topical Glucocorticoid Creams," Pharmacopeial Forum, 19, 5048-5059, 1993.3 h7 ~! H; M; a/ f( X& {
. B$ f2 i4 f: B3. Corbo, M., T. W. Schultz, G. K. Wong, and G. A. Van Buskirk, "Development and Validation of In Vitro Release Testing Methods for Semisolid Formulati**," Pharmaceutical Technology 17(9): 112-128, 1993.; b1 g4 W( u! K) a
) `- N4 C4 s5 h& h
4. Li, J. B. and P. C. Rahn, "Automated Dissolution Testing of Topical Drug Formulati** Using Franz Cells and HPLC Analysis," Pharmaceutical Technology 17(7):44-52, 1993.* }8 i; s+ r9 T. a
7 d& ` S0 K( T9 Z2 n/ m( `+ H8 E
5. Shah, V. P. and J. S. Elkins, "In Vitro Release from Corticosteroid Ointments," Journal of Pharmaceutical Sciences, 84:1139-1140, 1995.
- R6 e* k6 p$ |( F, A* g2 E
( f& Z' n. n. X, c9 H# D6. Zatz, J.L., "Drug Release from Semisolids: Effect of Membrane Permeability on Sensitivity to Product Parameters," Pharmaceutical Research 2:787-789, 1995.5 k5 x/ [: h8 _1 [% {& M
1 \0 S* u- g. h) d1 y/ ? $ `8 D7 t' w0 V; ~. }" o. B \
23
! D( z3 w, P- E
0 k( y/ H2 Q- W& b8 \" E# C m表 1 – 成份和组成) w' b: ^; c8 c5 \$ z8 \/ ]
! q' ~/ l4 Y4 K+ ?
| 级别 | 变更 | 试验资料 | 申报资料 |
| | | | |
| 1 | 去除或部分去除颜色、香味或味道 | 产品出厂要求 | 年度报告(包括长期稳定性数据 |
| | 一种辅料的变更量不得超过批准量 | 稳定性:首个生产批次的长期稳 | 的所有信息) |
| | 的5%,而且所有辅料变更量的添加效应 | 定性数据 | |
| | 总和≤5% | | |
| | 主要是一种单一化学实体(纯度≥ | | |
| | 95%)的赋形剂的供应商变更或者任何其 | | |
| | 他赋形剂的供应商或技术等级的变更 | | |
| | | | |
| 2 | 一种辅料的变更量为批准量的>5% | 产品出厂要求 | Changes being effected补充申请 |
| | 且≤10%,而且所有辅料包裹拉的添加效 | 生效批记录 | (包括加速稳定性数据的所有信息) |
| | 应总和≤10% | 稳定性:1批3个月加速稳定性数 | 年度报告(长期稳定性数据) |
| | 赋形剂供应商变更(不包括1级变 | 据和首批生产批长期稳定性数据 | |
| | 更) | 体外释放试验 | |
| | 赋形剂技术等级的变更 | | |
| | 如果药物为混悬液,则包括原料药 | | |
| | 颗径分布的变更 | | |
| | | | |
| 3 | 超出2级变更中记录范围的辅料的 | 产品出厂要求 | Prior approval supplement(包括加 |
| | 所有质量和数量变更 | 生效批记录 | 速稳定性数据的所有信息) |
| | 如果药物为混悬液,则包括原料药 | 稳定性: | 年度报告(长期稳定性数据) |
| | 晶型的变更 | 已获得重要信息:1批3个月加速 | |
| | | 稳定性数据,前3个生产批次的长期稳 | |
| | | 定性数据 | |
| | | 尚未获得重要信息:3批3个月加 | |
| | | 速稳定性数据,前3个生产批的长期稳 | |
| | | | |
2 b# w7 N, c! ~4 ~
8 D) r; t' S w8 q" n p241 p& G) `2 R5 S% O0 ?* e
file:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps26.pngfile:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps27.pngfile:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps28.pngfile:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps29.pngfile:///C:/Users/ADMINI~1/AppData/Local/Temp/ksohtml7484/wps30.png 定性数据) e" C( H/ N% `8 S7 A% y& i
6 N+ n5 I* ^9 P' s8 M体外释放试验(仅鼓励)' e4 {5 g7 M* i+ w: u
5 w- }4 i8 M& w$ P! H( W. Q
体内生物等效性试验3 f1 s1 s$ h# n# m
# s5 T2 B$ B/ {8 M! X- A
注: 其他信息见正文。
" M, @5 i7 k- W) K" a4 j
5 W: H: y: P8 X0 ]. L8 ~25
7 c1 ^& I* u4 o2 P6 U/ P% I% u
& M8 e0 Q/ s9 C/ z表 2 – 成份和组份 – 防腐剂9 f1 _- D. O4 w
- P" m; S+ x; ^" F) r2 P, i5 u
| 级别 | 变更 | 试验资料 | 申报资料 |
| | | | | |
| 1 | 变更量为防腐剂批准量的10% | 产品出厂要求 | 年度报告 | |
| | 或更低 | 测试防腐剂在规定的最低水平时的有效性 | | |
| | | | | |
| 2 | 变更量超过防腐剂已批准量的 | 产品出厂要求 | Changes | being effected |
| | 10%,最多变更20% | 测试防腐剂在规定的最低水平时的有效性 | 补充申请 | |
| | | | | |
| 3 | 变更量超过防腐剂已批准量的 | 产品出厂要求 | Prior | approval |
| | 20%(包括去除)或者使用不同的防 | 生效批记录 | supplement(包括加速稳定 |
| | 腐剂 | 测试防腐剂在规定的最低水平时的有效性 | 性数据的所有信息) |
| | | 对于新防腐剂:鉴定和测定的分析方法;验 | 年度报告(长期稳定性 |
| | | 证研究表明新防腐剂不会影响测试 | 数据) | |
| | | 稳定性:1批3个月加速稳定性数据和首批生 | | |
| | | 产批次长期稳定性数据 | | |
| | | | | |
| | | 注: 其他信息见正文。 | | |
3 b9 g6 c9 n, b: v2 \
/ Q/ ^; u; i' m- b
26' E) p$ ~* B. Q. F8 B& J Y- O
! v7 ^! c8 P2 c8 O+ ]. |: H' J
表 3 – 生产设备3 D( }, U! [' I+ L3 r/ ]
9 y! m! a# f0 ?# B( b4 _+ u( p| 级别 | 变更 | 试验资料 | 申报资料 | | |
| | | | | | |
| 1 | 转移成份的非自动化和非机械化 | 产品出厂要求 | 年度报告(包括长期稳定性数据 | | |
| | 设备变更为自动化或机械化设备 | 稳定性:首个生产批次的长期稳定 | 的所有信息) | | |
| | 选择设计和操作原理相同的设备 | 性数据 | | | |
| | | | | | |
| 2 | 不同设计或不同操作原理的设备 | 产品出厂要求 | Changes being effected | | |
| 补充申请 | | |
| | 混合设备类型,如高切应力变更 | 生效批记录 | (包括加速稳定性数据的所有信息) | | |
| | 为低切应力,或者相反 | 稳定性:已获得重要信息:1批3 | 年度报告(长期稳定性数据) | | |
| | | 个月加速稳定性数据,首个生产批次 | | | |
| | | 长期稳定性数据 | | | |
| | | 未获得重要信息:3批3个月加速稳 | | | |
| | | 定性数据,和前3个生产批长期稳定性 | | | |
| | | 数据 | | | |
| | | 体外释放试验 | | | |
| | | | | | |
4 C9 O! v, _6 J/ g: i注: 其他信息见正文。5 C' Y8 h; O6 l
. g2 p" e( X: `8 k: R+ w O+ O
8 v$ t( J- C R+ t' S y
274 \1 }. D/ ?1 Y
6 z3 O' m. w4 C+ ^, ]0 I$ \; X! x+ b
表 4 – 生产工艺
" k4 Q& D) T8 I , V) ?3 V9 }+ t; `7 b
| 级别 | 变更 | 试验资料 | 申报资料 |
| | | | |
| 1 | 已批准申请范围内的工艺变更 | 产品出厂要求 | 年度报告 |
| | 成份添加顺序的变更 | | |
| | | | |
| 2 | 已批准申请范围之外的工艺变 | 产品出厂要求 | Changes being effected补充申请 |
| | 更 | 生效批记录 | (包括加速稳定性数据的所有信息) |
| | 各相的混合工艺 | 稳定性:已获得重要信息:1批3个 | 年度报告(长期稳定性数据) |
| | | 月加速稳定性数据,和首个生产批次的 | |
| | | 长期稳定性数据 | |
| | | 未获得重要信息:3批3个月加速稳 | |
| | | 定性数据,和前三个生产批次的长期稳 | |
| | | 定性数据 | |
| | | 体外释放试验 | |
| | | | |
| 注: 其他信息见正文。 | | |
! E8 _3 s- i2 p0 y9 J0 V- } / T1 Q+ W [1 {) e9 D9 g% a4 h
28) m7 _0 C @+ v6 r; O2 Z
: z* ^8 G4 i. @6 d K, z表 5 – 批量 `' T& Z0 P! B5 h s9 m, X
' p: i5 e+ s. F) j2 ^
| 级别 | 变更 | 试验资料 | 申报资料 | |
| | | | | |
| 1 | 批量的变更不超过关键性临床 | 产品出厂要求 | 年度报告(包括长期稳定性数据 | |
| | 试验/生物学测试批次规模的10倍 | 生效批记录 | 的所有信息) | |
| | | 稳定性:首个生产批次长期稳定性 | | |
| | | 数据 | | |
| | | | | |
| 2 | 批量变更超过关键性临床试验/ | 产品出厂要求 | Changes being effected补充申请 | |
| | 生物学测试批次规模的10倍 | 生效批记录 | (包括加速稳定性数据的所有信息) | |
| | | 稳定性: 1批3个月加速稳定性数 | 年度报告(长期稳定性数据) | |
| | | 据,和首个生产批次的长期稳定性数据 | | |
| | | 体外释放试验 | | |
| | | | | |
5 d* K, M4 ^& s1 s5 [! O7 O, C注: 其他信息见正文。
7 c3 L$ }! X0 m, y8 y1 o5 x 8 K8 N, t7 K8 K: }8 L1 m- M
3 E/ F. G ^5 V9 `- Y# f# G5 \" k29
* Y9 V, f4 A) H0 m4 N: k
8 {1 N' ~- o' Y$ f0 l6 a表 6 – 生产产地变更+ L- y0 J% S( P" |
- P( G$ s; z9 m! y8 }8 W
| 级别 | 变更 | 试验资料 | 申报资料 |
| | | | |
| 1 | 在一个工厂内 | 产品出厂要求 | 年度报告 |
| | | | |
| 2 | 相邻厂区内,或相邻城市 | 产品出厂要求 | Changes being effected补充申请 |
| | 街区工厂间 | 生效的批记录 | 年度报告(长期稳定性数据) |
| | | 新场所的位置 | |
| | | 稳定性:与长期稳定性有关的首个生产 | |
| | | 批次 | |
| | | | |
| 3 | 不同厂区 | 产品出厂要求 | Changes being effected补充申请 |
| | 合同生产商 | 生效批记录 | (包括加速稳定性数据的所有信息) |
| | | 新场所的位置 | 年度报告(长期稳定性数据) |
| | | 稳定性:已获得重要信息:1批3个月加 | |
| | | 速稳定性数据,和前三个生产批次长期稳定 | |
| | | 性数据 | |
| | | 未获得重要信息:3批3个月加速稳定性 | |
| | | 数据,和前三个生产批次长期稳定性数据 | |
| | | 体外释放试验 | |
| | | | |
| 注: 其他信息见正文。 | | |
5 a! D0 v0 B1 x# ~
9 Z, c/ J; z( t; a5 u! r+ M( S% r30/ Q# r, U* N: i" T) J
; J. N- j0 \& B3 D
COMIS #0 g* U. k& B0 S. K4 |& W1 V3 Y/ U
$ B% x/ o* o1 ], X. G
Created: VShah: 1996
; f% t7 l: a D* M; x 9 E1 U, |" z( H; N
Reviewed: KRoberts: January 1997" d( T- ?! N- n/ s
- Z# ]" A2 s' s2 ]4 j6 wEdited: VShah: May 7, 19973 l# o6 Z% t. Q5 K. i
7 C0 v# ~* h& S, j* j6 T8 b( W
Reviewed: NSager: May 12, 1997
4 W! ]" o" @7 Y9 O& e" ^ / g4 k9 `) _/ Q0 g7 {+ K
Proofed: OVieira: May 14, 19971 o2 L- S9 ^! x e6 s9 Q8 j
6 h" v8 M0 O. q$ N
Reviewed: JAxelrad: May 19, 1997
/ R9 j# ^5 s! m+ @, z- e2 D6 G / g& K% z' v) G5 k6 |' [
Changes: NDerr: May 21, 1997
! [* W+ ~- K) _& g
, |/ P( Y6 M- k9 o4 qFinal Review: VShah: May 21, 1997. l3 [/ A0 p% K9 M- J
9 P! h% |4 Z8 J) s% p/ @) p& R
Final proof: NDerr:May 21, 19972 j# n( S: I- G4 d% m% l
+ Z2 M$ w) b. H/ Q3 c8 h+ ?
| 欢迎光临 药群论坛 (http://www.yaoqun.net/) |
Powered by Discuz! X3.2 |
 ! n' R& F& O: ^
! n' R& F& O: ^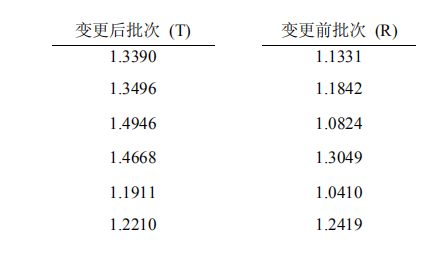 , V; _7 G% J+ F5 q
, V; _7 G% J+ F5 q