药群论坛
标题: 【BE审评】从法规解读到研究设计,CDE专家帮你划重点! [打印本页]
作者: xiaoxiao 时间: 2021-11-2 01:12 PM
标题: 【BE审评】从法规解读到研究设计,CDE专家帮你划重点!
我国是仿制药大国,在我国已有的18.9万个药品批准文号中,95%以上为仿制药,已成为制药行业的重要组成部分。在世界范围内,仿制药的供应也占据了ICH主要成员国药品市场的50%以上。. p/ h" b! M# I0 J( |1 r _
仿制药获批上市需满足一定的药学和临床要求,其中,关键的临床试验即为生物等效性(bioequivalence,BE)研究。另一方面,BE研究也广泛地应用于新药领域,可用于支持改良型新药、新药变更/新增规格、固定剂量复方等类型的注册申请。; | H, i, U! ` j4 B9 e! K
为什么BE研究可以作为关键研究桥接两种药物之间的安全性和有效性?
5 v: q D- ~/ v ]$ L; r 以仿制药为例,受试制剂和参比制剂具有相同且等量的活性成分,进入消化道后经过崩解、溶出、溶解等后透过胃肠壁吸收入血,随血液循环到达全身。( t3 x; r* I( O2 g# \5 U; b
在活性成分完全相同的情况下,如果其到达作用部位的速率和程度没有显著差异,那么产生的生物学效应理论上应是一致的。即通过药学一致、生物等效,可以推出治疗等效、具有临床可替代性。) }( P* W/ I Q7 C' ^* b% [. g1 h3 d* E
由此将BE定义为:在相似的试验条件下单次或多次服用相同剂量的试验药物后,受试制剂中药物的吸收速度和吸收程度与参比制剂的差异在可接受范围(±20%)内即为生物等效。4 z" R9 d+ z4 [
BE的研究和审评都要遵循特定的法规和指导原则。& z9 r/ M/ R8 I% P: c5 A* A' J
相关法规和工作文件包括《药品注册管理办法》(自2020年7月1日起实施)及其配套文件等;相关的指导原则包括:《中国药典》(2015年版通则9012生物样品定量分析方法验证指导原则)、2016年发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》以及《人体生物等效性试验豁免指导原则》等。
7 |5 n% w7 ]2 [* H' F 目前复杂制剂的BE研究要求也在积极推进中,如《化学药品注射剂仿制药(特殊注射剂)质量和疗效一致性评价技术要求》已于2020年发布,多项个药技术要求如《奥氮平口崩片生物等效性研究技术指导原则(征求意见稿)》等均在起草和征求意见中。
审评关注内容
. q: j5 U! Y* C% @& n$ k 1.参比制剂7 i/ M% Z7 M0 w' _# n
作为仿制药的标杆,参比制剂具有非常重要的地位。
/ S6 Y: P( I F+ ?$ o5 F, q 参比制剂一般要求为原研产品,且具有充分的临床安全有效数据。为避免误差传递,一般建议申请人选择国家药品监督管理局已发布的参比制剂开展BE研究。2 ?; g: H B0 i7 S; D# }
截至2020年9月30日,国家药品监督管理局已发布30批参比制剂,涉及约1585个品种及36448个品规,相关信息可在国家药品监督管理局药品审评中心(以下简称药审中心)网站一致性评价专栏或中国上市药品目录集中查询。
: E3 G$ U1 b1 t; H 2019年8月5日药审中心发布了《关于在审化学仿制药参比制剂有关事宜的通知》,要求对于注册分类3、4、5.2及化学仿制药一致性评价的品种,申请人在启动仿制药研究时,需尽早通过参比制剂申请平台提出遴选申请。
C& u) h* u2 G; e# w6 Z6 @ 若在审品种所选用的参比制剂不在国家药品监督管理局已发布的目录中,也需尽快提出申请。9 M, m& v# c4 ?# c( B7 k: m
2.研究设计
# @/ d5 a& y( ^7 p* @ BE的研究设计包含多方面的内容,如研究类型、受试者、给药剂量、检测对象及评价指标等。
5 i5 C k O$ O( f9 ]( J$ z 一般的BE都是两制剂、单次给药、交叉试验设计;对于长半衰期药物可以选择平行设计,但需关注组间均衡性问题;而高变异药物可考虑重复设计。
( Q" C+ F" L0 h9 H7 |) Z 研究人群一般是纳入健康受试者,但当健康受试者可能面临安全性方面的风险时,建议选择适应证患者人群,此时一般设计为多次给药。
, z) ~" ` C( o! l5 v# j6 G( O, ?5 q 2005年《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》推荐给药剂量采用临床单次用药剂量,但2016年《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》已对此进行修订,建议采用申报最高规格,以一个单位(如单片或单粒)服药。
) \+ s+ V4 V% c1 N 测定对象一般是原型药物,因为原型药物的药时曲线比代谢产物能更灵敏地反映制剂间的差异。
5 d5 l; H8 o) d9 |& l+ P+ b 总体来说,试验设计需结合品种药动学特点、剂型、作用机制等综合评估其合理性。9 y" c. Q9 p5 g) s
3.研究实施
) z; T0 w" o) B! T% d, y BE研究的实施可分为两部分:
7 @) r' ?( t1 t1 g( o9 W8 R4 L ①临床实施。" P3 ^. @; c& u) m! n# E0 B
临床试验的管理需符合伦理和药物临床试验质量管理规范(GCP)的要求,需关注两周期过程标准化、受试者管理、血样采集、服药过程、药品及样品的管理以及方案偏离的处理等关键环节。; Y/ r, K Z" ^* N
②方法学验证及生物样品分析。/ Y% R% M; ]" E2 o" O- V
临床试验所用的生物药品大多为血样,在进行分析之前须进行充分、全面的方法学验证,包括标准曲线、精密度、准确度、选择性、基质效应、回收率及稳定性等。生物样品分析主要关注复测是否遵循标准操作程序(SOP)的要求、复测是否合理、是否有手动积分以及试验样品再分析(ISR)结果和相关调查等。# m1 Q% d. A5 t2 ?: |5 n6 N0 U
FDA总结了2001~2011年4028项简略新药申请(ANDA)的分析缺陷,其中,62%为方法学验证的问题,35%是分析报告的缺陷,3%为分析方法存在的问题,比较突出的包括长期稳定性不充分、原始数据未充分提交、色谱图不完整等。: K8 r+ H+ g' X1 K( o4 l8 A& `
研究过程实施的规范性和数据可靠性是审评的基础,这也是现场核查的主要关注内容。以审评为主导,核查、检验等为支撑的药品注册体系正在建立中。$ H/ I- X+ ]# o' Z# O% A1 t
按照新的药品注册管理办法配套文件的要求,2020年药审中心发布了《药品注册核查检验启动原则和程序管理规定(试行)》的征求意见稿,针对注册核查由以往的“逢审必查”转变为“基于风险启动核查”,同时将审评与检查、检验工作由“串联”调整为“并联”。根据品种因素和研发生产主体合规风险因素,经综合评估研判后,对注册申请进行风险等级划分。
+ K! j6 w7 Q! V5 \: J# @4 [6 U! b+ }2 T 对于高风险级别的注册申请,原则上应全部启动核查;中、低风险的按不同比例启动核查。这种基于风险理念的核查办法和“并联”模式的改变,加强了高风险机构的监督检查以防范系统性风险,节约了行政资源,提高了审评效率,有效地保证了数据的可靠性。$ I0 K k/ l5 U4 d1 {- b
4.结果评价
8 u3 b3 x. |" B5 y, [+ X 对研究结果的评价主要关注以下方面:4 G* I3 U+ k0 r$ t1 Y5 B5 S4 p) {; F
①数据集纳入是否合理:包括受试者的入组、脱落、剔除和异常值的处理等。
4 H5 K& T( s7 y9 l% N ②统计结果是否符合要求:药物浓度-时间曲线下面积(AUC)和最大血药浓度(Cmax)几何均值比值的90%置信区间是否在80.00%~125.00%之间;受试制剂的安全风险情况。+ A7 o. Z8 F7 J: [9 n
③方案偏离的处理:对采血点超窗、合并用药等情况的处理及评估等。
特定药物BE审评和案例分析
9 H" G) E) F- M, e7 c 1.特定剂型及给药方式6 ]# [& X4 [, A5 R3 a$ J
不同剂型可能存在不同的给药方式,BE试验过程中需关注特定剂型及给药方式的特点。
. @; ` ^" h, e 如昂丹司琼口溶膜,一般给药方式为整片放于舌,溶解后随唾液吞下,临床实施过程中应注意记录受试者的口感、刺激性和砂砾感;
D4 V4 B' e4 a- K9 l) J4 y 尼古丁咀嚼胶,按照说明书用法咀嚼服用30min,完成咀嚼后,还需测定咀嚼胶残留量;
' [3 g/ A G. v' z. T5 R+ s/ V: O 美沙拉秦灌肠液,给药方式为直肠给药,应关注受试者给药前的排空及直肠保留依从性评估等。$ }& ]% v7 }. L% Q
对于吸入制剂、透皮贴剂等其他复杂制剂的技术要求也正在研究讨论中。9 T4 [3 h# M5 ]7 _
2.特定研究方法
4 q' v5 i; E' E) Z( z# t H1 b BE的研究方法包括药动学(PK)、药效学(PD)、临床研究或体外研究,其评价效力不同。
" n6 ^2 ], Q7 Z" K% U9 Z0 d PK是首选也是最常用的研究方法。在PK法不适用时,才会考虑PD或临床研究方法。
3 M* H$ ]1 ]. C/ R4 D 体外方法仅适用于特殊情况,如磷结合剂,为胃肠道局部作用药物,口服不吸收,无法用PK法进行BE评价。以碳酸镧为例,其生物利用度低于0.002%,服药后在消化道上端酸性环境中解离释放镧离子,与食物中磷结合沉淀,通过减少磷吸收从而降低患者的血磷,用于治疗透析患者高磷血症。这种情况下,可考虑采用PD法或体外法进行评价。
1 I5 |0 [$ H( F/ U PD法是通过尿磷排泄反映外源性磷的吸收,进而反映药物制剂的作用;体外法是通过模拟体内的环境,考察在不同pH条件下的溶出曲线及相似因子(f2)是否一致,评估制剂的药学特性。同时,通过磷动力学结合试验与磷平衡结合试验分别确定孵育时间、测定亲和力常数k1和结合能力常数k2,比较两者结合曲线的差异,从而反映药效的差异。+ c* g9 v) P# Y4 A( ~4 X3 c7 f
3.特定评价标准8 j* g" I0 b( Y! T8 D
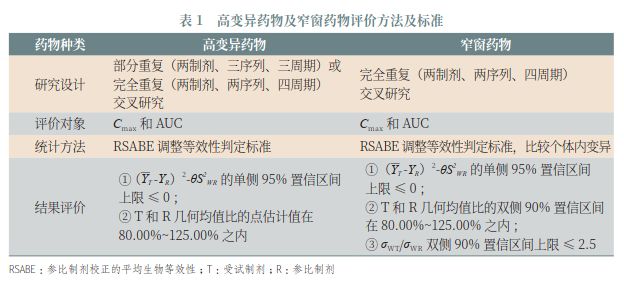
对于高变异药物及窄窗药物(如卡马西平、环孢素)等在研究设计和评价标准上也可能需要做出相应的调整(表1)。) R! V3 @$ w9 W
4. 审评案例分析:泮托拉唑钠肠溶片
% s: Y n2 j; i! s/ @9 h1 ~7 t: U 质子泵抑制剂如泮托拉唑钠肠溶片的餐后BE研究常常出现问题。9 ?7 @$ I3 d/ B* o$ G* P
例如,在申报资料中,会出现血药浓度极低甚至为0的受试者。由于无法区分这种现象是否由制剂因素造成,不能直接剔除异常值进行BE评价。并且,指导原则中明确要求,从统计分析中排除一名受试者的决定必须在生物分析之前做出,不能接受基于统计分析或单纯的药动学理由排除数据。3 }9 H6 r6 ]/ i, B9 P7 y( y5 K" ~
因此,需要多角度分析这类情况出现的可能原因:如个体变异高、受试者吐药、肠溶包衣在胃中释放导致药物在酸中不稳定或者未完全崩解仅部分溶出等。排除上述可能性后,再进一步分析BE的试验设计中可能存在的问题。
8 q0 a, b; p2 w6 j& W3 P( Y- q 某些情况下,肠溶制剂的胃排空可能会延长且高度不稳定,可能是由胃肠道移行性复合运动(migrating motor complex,MMC)引发。
6 b( K8 m: k1 ~+ ~ Z" [2 K6 G- K MMC通常发生于两餐之间,为间歇性活动,有研究认为其目的是在空腹时清除胃和小肠的食糜,将两餐之间残存食物推进至下一段消化道。如果肠溶制剂在胃内长时间滞留后才被排空,同时BE采血点设计在药时曲线后期较为稀疏,可能会捕捉不到吸收延迟受试者药物的真实暴露情况。
# `( J* U9 \. x& ^ 因此,对于此类药物,在设计采血点时不仅需要考虑药物的半衰期,还需考虑该效应的可能性,否则会导致异常值出现及BE不等效。
结语
4 |1 j, @. m! l/ b BE审评是在遵循法规和指导原则的基础上,结合药物作用机制、制剂特点、生理过程、统计学方法等综合评价的一门学科。
4 |, n9 y. E8 E; _3 q5 @ 随着仿制药的日益复杂,技术审评也面临着越来越多的挑战,需要不断研究新的评价方法和要求。* F+ j$ c( I0 Z) X, E; O1 {
2017年中国加入ICH,标志着我国将逐步与国际高技术标准和要求同步,参与国际规则的制订,对我国技术审评提出了更高的要求。只有不忘初心,持续学习和研究,不断优化和完善已有的指导原则体系,才能实现更快、更好地把高品质的仿制药推向临床的目标。3 h# {0 ^! Y: T% W3 D% _
文章来源:《中国食品药品监管》9 A) I& I$ e( G6 P/ \% q# R
原标题:《生物等效性研究的审评考虑》
9 v8 \1 O/ l9 x) j6 f 作者:李敏,李芳,杨进波(国家药品监督管理局药品审评中心)/ b; ?' @& G; R
. j; a- J8 ^/ z& l
作者: lantianbaiyun20 时间: 2021-11-5 08:01 AM
8 ?/ i8 x0 N) Z3 F E
6 f4 O' g* O" U6 w& G谢谢分享,拜读了
作者: luyuebing 时间: 2022-3-18 01:53 PM
谢谢分享,学习一下
作者: Irene123 时间: 2022-10-18 09:06 AM
谢谢分享
| 欢迎光临 药群论坛 (http://www.yaoqun.net/) |
Powered by Discuz! X3.2 |
 ' W1 k4 `" _9 C, ?+ D; Z4 T
' W1 k4 `" _9 C, ?+ D; Z4 T